Гематология-Миелодиспластический синдром
Миелодиспластический синдром
URL
FONT face=Arial size=2>
В конце 30-х, начале 40-х годов ХХ столетия внимание исследователей привлекла группа заболеваний проявляющихся анемией, рефрактерной к какой-либо терапии. Больные с такой патологией не отвечали на лечение препаратами железа, витаминами группы В и фолиевой кислотой, что дало основание использовать для обозначения данной болезни термина "рефрактерная анемия". В отечественной литературе эти заболевания обычно рассматривылись под термином "ахрестические анемии".
Наблюдение за больными с рефрактерной анемией позволило выявить в ряде случаев феномен увеличения количества бластных клеток в костном мозге до 30 %. Таких больных стали обособлять в группу с названием "предлейкоз", "малопроцентный лейкоз" или "дремлющий лейкоз ". Отграничивая их тем самым от хорошо известных заболеваний костного мозга, протекающих с высоким (больше или равно 30%) бластозом костного мозга, -- острых лейкозов. У некоторых больных данной группы в последующем развивался острый лейкоз (количество бластов в костном мозге превышало 30 %).
Дальнейшие исследования позволили выявили у больных с резистентной к терапии анемией, как с увеличением количества бластных клеток в костном мозге, так и без, ряд особенностей. Эти особенности касались в первую очередь морфологии костного мозга (нарушение архитектоники ростков гемопоэза, изменения стромальной ткани, признаки дисплазии кроветворных клеток). Кроме того, при выполнении цитогенетического исследования у таких больных часто выявлялись изменение кариотипа гемопоэтических клеток.
Цитогенетическими и ферментными методами была доказана клональная (опухолевая) природа заболевания. Характерной особеностью клеток, происходящих из опухолевого клона, при миелодиспластическом синдроме является их морфологическая и функциональная неполноценность.
Дальнейшие наблюдения позволили установить, что миелодиспластический синдром является неоднородной группой. У больных миелодиспластическим синдромом отмечались существенные различия в тяжести клинических проявлений, длительности течения заболевания (сроках выживаемости больных), в частоте и скорости озлокачествления заболевания -- трансформации в острый лейкоз. Различия в течение заболевания требовали и различной тактики ведения больных, то есть возникла необходимость выделения самостоятельных нозологических форм.
Для обозначения данной группы заболеваний был принят термин "миелодиспластический синдром" в рамках которого были описаны пять самостоятельных нозологических форм (заболеваний).
Миелодиспластический синдром (МДС) -- это группа заболеваний костного мозга, носящих клональный характер и возникающих в результате мутации стволовой клетки крови. При этом потомки мутировавшей стволовой клетки сохраняют способность к дифференцировке до зрелых клеток. Однако процесс дифференцировки носит неэффективный характер, в результате чего зрелые клетки крови изменены морфологически, уменьшены в количестве и ослаблены в функции.
Сегодня общепринятой является классификация МДС разработанная Франко-Америко-Британской исследовательской группой (FAB) и опубликованная в 1982 году. В основе классификации лежат четыре признака:
-- количество бластов в костном мозге;
-- количество бластов в периферической крови;
-- количество атипичных (кольцевидных) сидеробластов в костном мозге;
-- количество моноцитов в периферической крови.
Такой набор признаков позволяет разделить группу схожих заболеваний на самостоятельные нозологические формы (таблица 1), которые отличаются по частоте встречаемости, длительности течения, вероятности озлокачествления (трансформации в острый лейкоз) и требуют различной тактики лечения больных. Частота встречаемости различных заболеваний, выделяемых в группе МДС, длительность выживания больных и вероятность трансформации в острый лейкоз представлены в таблице 2.
Таблица 1. FAB-классификация миелодиспластического синдрома (по Bennett et al., 1982).
1.Рефрактерная анемия:
а) количество бластов в костном мозге менее 5%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
2.Рефрактерная анемия с кольцевыми сидеробластами:
а) количество бластов в костном мозге менее 5%
б) количество кольцевых сидеробластов в костном мозге не менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
3.Рефрактерная анемия с избытком бластов:
а) количество бластов в костном мозге более 5%, но менее 20%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 5%
г) количество моноцитов в периферической крови менее 1х10^9/л
4.Рефрактерная анемия с избытком бластов на стадии трансформации:
а) количество бластов в костном мозге более 20%, но менее 30%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
5.Хронический миеломоноцитарный лейкоз:
а) количество бластов в костном мозге менее 20%
б) количество кольцевых сидеробластов в костном мозге любое
в) количество бластов в периферической крови менее 5%
г) количество моноцитов в периферической крови не менее 1х10^9/л
Таблица 2. Частота встречаемости каждого из нозологических вариантов МДС, длительность выживания и вероятность трансформации в острый лейкоз.
частота (%) выживаемость (мес) вероятность(%)
1.Рефрактерная анемия: 25 37 11
2.Рефрактерная анемия с кольцевыми сидеробластами: 18 49 5
3.Рефрактерная анемия с избытком бластов: 28 9 23
4.Рефрактерная анемия с избытком бластов
на стадии трансформации: 12 6 48
5.Хронический миеломоноцитарный лейкоз: 17 22 20
Эпидемиология. МДС -- патология старшей возрастной группы. 80% случаев МДС приходится на лиц старше 60 лет. В европейских странах среди лиц 50 -- 69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше -- 150 новых случаев на 1 млн населения.
Этиология. Несмотря на многочисленные исследования, причины вызывающие развитие МДС остаются во многом неясными. В группе этиологических факторов рассматривают факторы, способные вызывать мутации клеток и тем самым приводить к развитию опухоли: вирусы, ионизирующее излучение, химические агенты. На сегодняшний день каких-либо этиологических факторов, специфичных для МДС не установлено. В ряде случаев развитию МДС предшествует химиотерапия солидных опухолей.
Патогенез. Отправной точкой в развитии МДС является мутация стволовой клетки крови. Потомки мутировавшей клетки получают биологическое преимущество перед нормальными гемопоэтическими клетками, что позволяет им полностью колонизировать костный мозг, вытесняя нормальные гемопоэтические клетки. Особенностью мутации стволовой клетки крови при МДС является частичное сохранение ее потомками способности к созреванию до зрелых клеток крови. Однако, процесс созревания носит неэффективный характер, что приводит к уменьшению количества зрелых клеток в периферической крови. Кроме количественных изменений в составе клеток периферической крови имеет место и снижение их функциональной активности.
Немаловажную роль в развитии патологического клона гемопоэтических клеток играет стромальное микроокружение, однако конкретные механизмы вовлечение стромальной ткани в патологический процесс при МДС изучены еще недостаточно.
Неэффективный характер гемопоэза (дисплазия кроветворения) имеет хорошо выраженный морфологический эквивалент -- изменение как морфологических признаков гемопоэтических клеток, так и их расположения внутри костномозговой полости (изменение архитектоники). Третью составляющую морфологических признаков дисплазии кроветворения образуют изменения стромальной ткани. Основные морфологические признаки дисплазии дисплазии кроветворения представлены в таблице 4 и таблице 5.
Таблица 4. Морфологические признаки дисплазии кроветворения при исследовании аспирата костного мозга (по Bartl R, Frisch B и Baumgart R, 1992).
Эритроидная линия:
-- эритроидная гиперплазия
-- мегалобластоидность
-- многоядерность
-- фрагментация ядер
-- межядерные мостики
-- вакуолизация цитоплазмы
-- PAS-позитивные нормобласты
-- кольцевые сидеробласты
Мегакариоцитарная линия:
-- микромегакариоциты
-- большие мегакариоциты с одним или несколькими мелкими круглыми ядрами
-- митотические фигуры
-- пикноз
-- гигантские тромбоциты
Гранулоцитарная линия:
-- гранулоцитарная гиперплазия
-- увеличение бластных клеток
-- парамиелоидные клетки
-- палочки Ауэра
-- гипо- и гипергранулярность
-- Пельгеровская аномалия
-- базофилия цитоплазмы зрелых клеток
-- эозинофилы с кольцевыми ядрами
Моноцитарная линия:
-- моноциты с множественными вытянутыми лопастями цитоплазмы
-- афзурофильные гранулы в цитоплазме
-- гемофагоцитоз
-- железосодержащие макрофаги
Ниже представлены несколько фотографий, иллюстрирующих морфологические признаки дисплазии кроветворения.
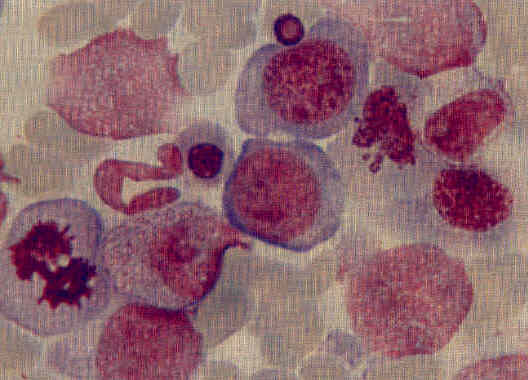
Рисунок 1. Дисплазия эритроидного ростка в костном мозге: мегалобластоидность, асинхронность ядер, тельца Жоли.
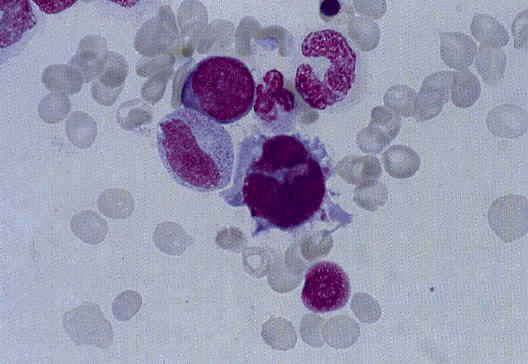
Рисунок 2. Дисплазия мегакариоцитарного ростка в костном мозге: микромегакариоцит.
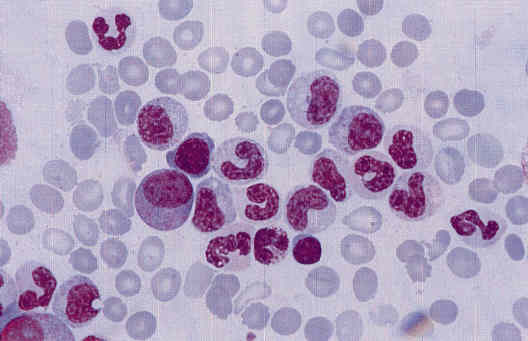
Рисунок 3. Дисплазия гранулоцитарного ростка в костном мозге: значительная редукция числа гранул.

Рисунок 4. Костный мозг больного рефрактерной сидеробластной анемией: кольцевые сидеробласты.
Таблица 5. Гистологические признаки дисплазии кроветворения при исследовании биоптата костного мозга (по Bartl R, Frisch B и Baumgart R, 1992).
Клеточность костного мозга:
-- гиперклеточный (свыше 50% случаев)
-- нормоклеточный (30-40% случаев)
-- гипоклеточный (менее 20% случаев)
Гистотопография:
-- атипичная локализация незрелых предшественников
-- атипичная локализация эритроцитарных предшественников
-- атипичная локализация мегакариоцитов
-- интраваскулярное расположение гемопоэтических клеток
Стромальные изменения:
-- экстравазация эритроцитов
-- разрывы синусоидов
-- расширение синусоидов со склерозом стенок
-- интростициальный и парамегакариоцитарный фиброз
-- лимфоидные узелки
-- плазмацитоз
-- лимфоцитоз
-- увеличение тучных клеток
-- увеличение костного преобразования
В неопластическом клоне могут происходить вторичные мутации клеток. В ряде случаев это приводит к развитию в костном мозге клона гемопоэтических клеток, потерявших способность к созреванию в большей степени, чем гемопоэтические клетки из предшествующего клона. Морфологическим эквивалентом этого события является увеличение в костном мозге количества незрелых клеток -- бластов. Если количество бластов в цитологическом препарате костного мозга превышает 30%, то говорят о трансформации в острый лейкоз. При этом следует понимать, что данная ситуация не означает развитие нового (второго) заболевания, а является закономерным продолжением течения данной нозологической фомы миелодиспластического синдрома, подчиняющегося закону опухолевой прогрессии. Трансформация в острый лейкоз при МДС эквивалентна развитию бластного криза при хроническом миелолейкозе.
Следует сделать отступление и пояснить, что в данной ситуации мы встречаемся с терминологическими сложностями, которые очень характерны для гематологии. Под термином "острый лейкоз" скрываются два неидентичных понятия. Первое понятие -- острый лейкоз как самостоятельное, первично возникающее заболевания костного мозга для которого характерна полная утрата опухолевыми клетками способности к созреванию. Второе понятие -- острый лейкоз как стадия развития миелодиспластического синдрома, когда опухолевые клетки первоначально сохраняют способность дифференцироваться до зрелых клеток крови и утрачивают эту способность только после повторных мутаций. Чтобы избегать возможного недопонимание предложено использовать для обозначения первой ситуации (самостоятельно возникший острый лейкоз) термин "de novo острый лейкоз", а для обозначения второй ситуации - термин "острый лейкоз развившийся (трансформировавшийся) из предшествующего миелодиспластического синдрома". Развитие острого лейкоза не является обязательной стадией в течении МДС. Частота наступления этого события различна у заболеваний образующих группу МДС и зависит от степени утраты опухолевыми клетками способности созревать в дебюте заболевания.
Таким образом, основным патогенетическим событием при МДС является возникновение в результате мутации стволовой клетки крови неопластического клона с частично нарушенной способностью к созреванию. Опухолевый клон вытесняет из костного мозга нормальные гемопоэтические клетки, и гемопоэз в костном мозге осуществляется только потомками мутировавшей клетки. В результате описанных процессов зрелые клетки крови имеют опухолевой происхождение, уменьшены в количестве и ослаблены в функции. С течением времени в опухолевых клетках могут произойти вторичные мутации, что приводит к полной утрате этими клетками способности созревать -- наступает конечная фаза развития МДС для обозначения который используют термин "острый лейкоз развившийся из предшествующего МДС".
Клиническая картина. Клиническая картина при различных формах МДС схожа и во многом определяется показателями периферической крови. Изменения периферической крови прямо зависят от степени нарушения созревания гемопоэтических клеток. Анемия постоянный и обязательный признак. Для нее характерны гиперхромия (высокий цветовой показатель) и макроцитоз. Уровень снижения гемоглобина может варьировать от умеренного до значительного. От степени и скорости нарастания анемии будет зависеть самочувствие больного. При медленном снижении гемоглобина организм успевает адаптироваться к гипоксии и количество жалоб у больных может быть минимальным. Если анемия развивается быстро, больные предъявляют жалобы на общую слабость, утомляемость, сердцебиение, одышку. Может утяжеляться течение ишемической болезни сердца, появляются признаки сердечной недостаточности.
Снижение количества зрелых гранулоцитов (нейтропения), а также их функциональная несостоятельность влекут за собой инфекционные осложнения. У 10 % больных развиваются стоматиты, гингивиты, пневмонии, инфекция мочевыводящих путей, абсцессы различной локализации, сепсис. У 20 % больных данной группы инфекционные осложнения становятся причиной смерти. Наиболее многочислены осложнения бактериальной природы, возбудителями которых являются Escherichia coli, Pseudomonas pyocyanea, Klebsiella pneumoniae, Staphylococcus aureus и Streptococcus fecalis. Также достаточно часто тяжелые инфекционные осложнения вызываются Pneumocystic carinii, Cryptococcus neoformous, Candida albicaus, Aspergillus fumigatus и цитомегаловирусом, что связано с функциональной неполноценностью Т-лимфоцитов при МДС.
Клинически значимая тромбоцитопения (приводящая к развитию геморрагического диатеза с петехиально-пятнистым типом кровоточивости) встречается у 15 % больных МДС. У половины из них кровотечение или кровоизлияния становятся причиной смерти. В некоторых случаях МДС, как правило у больных рефрактерной анемией, может отмечаться тромбоцитоз. Проявления гиперпластического синдрома в виде спленомегалии, гепатомегалии, лимфоаденопатии и специфического поражения кожи (лейкемиды) имеют место в основном у больных ХММЛ.
Спленомегалия встречается у 17 % таких больных, гепатомегалия у 13 %, а лейкемиды у 10 %.
Диагностика.Отправной точкой диагностического поиска являются, как правило, жалобы связанные со снижением уровня гемоглобина, подкрепляемые выявлением гиперхромной, макроцитарной анемии при исследовании периферической крови. Выявление при первичном осмотре, наряду с анемическими жалобами, явлений геморрагического диатеза и/или гиперпластического синдрома позволяют сформировать представление о
патологии системы крови еще до получения результатов лабораторных исследований. Наличие би- или трицитопении в периферической крови является абсолютным показанием для морфологического исследования костного мозга. При исследовании аспирата костного мозга у больных МДС в большинстве случаев определяется гиперплазия всех ростков кроветворения и обязательно выявляются признаки дисплазии клеток (смотри "Миелодиспластический синдром. Часть 1"). Максимальную информативность имеет гистологическое исследование костного мозга, получаемого методом трепанобиопсии. Гистологическое исследование позволяет обнаружить высоко специфичную для МДС морфологическую картину.
В качестве вспомогательного метода диагностики может быть использовано цитогенетическое исследование кариотипа гемопоэтических клеток. Различные хромосомные поломки выявляются у 48 % больных МДС. Частота аномалий кариотипа варьирует в зависимости от нозологического варианта МДС. Так у больных РА хромосомные поломки обнаруживаются в 30 % случаев, а у больных РАИБтранс в 60 %. Выявление хромосомных аномалий имеет большое значение для определения прогноза течения заболевания.
Наиболее часто встречающимся изменением кариотипа у больных РА является делеция (утрата) части длинного плеча пятой хромосомы (5q-). Данная аномалия чаще выявляется у женщин (соотношение мужчин и женщин среди заболевших составляет 1:5). Для больных с такой хромосомной поломкой характерны ярко выраженые морфологические аномалии мегакариоцитов (микромегакариоциты), тромбоцитоз периферической крови и достаточно благоприятное течение заболевания с низкой частотой трансформации в острый лейкоз.
Диагноз складывается из морфологически подтвержденного представления о наличии у больного миелодиспластического синдрома и окончательно формулируется (нозологическая форма) на основании количественных критериев миелограммы и гемограммы (FAB-классификация):
РА - бласты костного мозга < 5%, кольцевые сидеробласты <15%, бласты периферической крови < 1%, моноциты периферической крови < 1x10^9/л;
РСА - бласты костного мозга < 5%, кольцевые сидеробласты >15%, бласты периферической крови < 1%, моноциты периферической крови < 1x10^9/л;
РАИБ - бласты костного мозга 5 -20%, кольцевые сидеробласты - любое количество , бласты периферической крови < 5%, моноциты периферической крови < 1x10^9/л;
РАИБТ - бласты костного мозга 20-30%, кольцевые сидеробласты - любое количество , бласты периферической крови > 5%, моноциты периферической крови < 1x10^9/л;
ХММЛ - бласты костного мозга < 20%, кольцевые сидеробласты - любое количество, бласты периферической крови < 1%, моноциты периферической крови > 1x10^9/л;
Дифференциальный диагноз. Направление дифференциально-диагностического поиска будет зависеть от характера изменений гемограммы. Выявление гиперхромной, макроцитарной анемии как моносимптома или в сочетании с нейтропенией и/или тромбоцитопенией делает необходимым проведение дифференциального диагноза МДС с:
-- витамин В12-дефицитной анемией;
-- фолиеводефицитной анемией;
-- апластической анемией;
-- пароксизмальной ночной гемоглобинурии;
-- гипопластическим и алейкемическим вариантами острого лейкоза;
-- метастазами рака в костный мозг.
При выявлении в анализе периферической крови би- или трицитопении, бластов, промиелоцитов и миелоцитов, является абсолютным показанием для гистологического исследования костного мозга, сводящего вопрос о дифференциальном диагнозе к минимуму.
Лечение. Для лечение больных рефрактерной анемией (РА) используется заместительно-трансфузионная и симптоматическая терапия. Зависимость от трансфузий эритроцитарной массы может колебаться в широких пределах. При наличии глубокой тромбоцитопении и/или тяжелых проявлений геморрагического диатеза показаны трансфузии тромбоцитарной массы.
Длительная заместительная терапия эритроцитарной массой со временем приводит к избыточному накоплению железа в организме. В связи с этим, больным необходимо мониторировать уровень сывороточного железа. При увеличении его содержания свыше 30 мкмоль/л необходимо вводить десферал для профилактики гемосидероза внутренних органов.
Химиотерапия больным РА не проводится. Применение глюкокортикоидов, андрогенов, анаболических препаратов иногда имеет положительный эффект, но длительность выживания больных не возрастает. Кроме того, доказано, что при лечении препаратами этих групп увеличивается частота трансформации в острый лейкоз.
Рефрактерная анемия с кольцевыми сидеробластами (РАС) является самой доброкачественной формой МДС. Зависимость от гемотрансфузий у больных РАС невысока. В другой терапии они, как правило, не нуждаются и длительно сохраняют высокое качество жизни.
Единственным методом лечения больных РАИБ и РАИБтранс, позволяющим рассчитывать на излечение является аллогенная трансплантация костного мозга. При отсутствии донора, больным РАИБ и РАИБтранс показана химиотерапия. Лечение цитостатиками начинают у больных с количеством бластов в костном мозге свыше 10%. Применение интенсивной химиотерапии позволяет добиться ремиссии у 20-30 % больных, но, к сожалению, длительность ремиссии не превышает в среднем двух месяцев и не влияет на общую продолжительность жизни больного. В качестве базисного препарата для химиотерапии используется цитозар. Применяется также этопозид и рубомицин. Дозы и схемы введения различны. Проведение химиотерапии позволяет уменьшить зависимость больных от гемотрансфузий и продлить длительность жизни.
Больные ХММЛ в дебюте заболевания нуждаются только в трансфузионной и симптоматической терапии. При увеличении количества бластов возникают показания к цитостатической терапии. Базисным препаратом для проведения химиотерапии у больных ХММЛ является этопозид.
При развитии острого лейкоза у больных любой формой МДС как правило проводится полихимиотерапия принятая для данного варианта острого лейкоза. Следует отметить, что острый лейкоз, развившийся из предшествующего МДС, плохо поддается терапии. Ремиссию удается получить редко и она не продолжительна.
Прогноз. Длительность выживания больных РА и РАС составляет в среднем 3 - 4 года. При адекватной трансфузионной терапии больные достаточно длительный период времени сохраняют хорошее качество жизни и могут быть трудоспособны. Прогноз у больных ХММЛ несколько хуже, а у больных РАИБ и РАИБтранс плохой. Длительность выживания больных при трансформации любой из нозологических форм МДС в острый лейкоз ограничивается 4 -- 6 месяцами.