Терапия-Нейрогормоны и цитокины при сердечной недостаточности: новая теория старого заболевания?
Нейрогормоны и цитокины при сердечной недостаточности: новая теория старого заболевания?
Беленков Ю.Н., Агеев Ф.Т., Мареев В.Ю.
НИИ кардиологии им. А.Л.Мясникова РК НПК Минздрава РФ, Москва
URL
Резюме
Реальная практика лечения больных ХСН показывает, что ингибиторы
АПФ вместо ожидаемых 100% позволяют добиться снижения риска смерти
в среднем всего лишь на 23%. Одной из причин "низкой" эффективности
ИАПФ может явиться то, что наряду с нейрогормонами центральную
роль в патогенезе заболевания играют провоспалительные цитокины:
ФНО-a, ИЛ-1 и ИЛ-6. Механизм действия
цитокинов при ХСН складывается из отрицательного инотропного действия;
ремоделирования сердца, нарушения эндотелий-зависимой дилатации
артериол и усиления процесса апоптоза кардиомиоцитов. Источником
избытка цитокинов при сердечной недостаточности могут быть "перенапряженные"
кардиомиоциты или клетки периферической мускулатуры; однако есть
данные, что выработку цитокинов провоцируют эндотоксины, проникающие
в организм больного ХСН через отечную стенку кишечника. "Цитокиновая"
модель патогенеза предполагает возможность использования у больных
ХСН препаратов, блокирующих синтез ФНО-a или ингибирующих активность
ФНО-a.
Среди последних наиболее перспективным является энтерасепт - препарат,
представляющий собой фрагмент растворимого рецептора к ФНО-a, первый опыт использования которого
при ХСН дал обнадеживающие результаты. Возможно, что одним из
механизмов положительного действия ингибиторов АПФ у больных ХСН
также является их способность воздействовать на синтез цитокинов.
Neurohormones and cytokines in heart failure: a new theory for the old disease?
Belenkov Yu.N., Ageyev F.T., Mareyev V.Yu.
Summary
Actual practice of managing patients
with chronic heart failure (CHF) shows that ACE inhibitors provide
a mean reduction of the risk for death only by 23% instead of
100% expected. One reason for such a "low" efficacy of ACE inhibitors
may be the fact that along with neurohormones, proinflammatory
cytokines, TNF-a, IL-1 and IL-6 play a crucial role in the pathogenesis of
the disease. The mechanism of cytokine action in CHF involves
a negative inotropic effect; heart remodeling; impaired endothelium-dependent
dilation of arterioles; and enhanced apoptosis of cardiomyocytes.
The excess of cytokines can originate from "overstrained" cardiomyocytes
or peripheral muscle cells. However, there is evidence that cytokine
production can be provoked by the endotoxins penetrating into
the body of a patient with CHF through edematous intestinal wall.
The "cytokine" pathogenetic model suggests a possibility of treating
CHF patients with the agents that block TNF-a
synthesis or inhibit TNF-a activity.
Of the latter, enteracept soluble TNF-a
receptor a fragment is the most promising. Early experience of
with the drug in CHF has provided encouraging results. The capability
of ACE inhibitors for influencing the cytokine synthesis is possible
one of the mechanisms underlying the beneficial effect of these
drugs in patients with CHF.
Введение
Несмотря на всю убедительность современной
нейрогуморальной теории, согласно которой основной причиной развития
синдрома хронической сердечной недостаточности (ХСН) является
гиперактивация ренин-ангиотензин-альдостероновой (РААС) и симпатоадреналовой
систем, в последние годы появляется все больше клинических фактов,
которые невозможно объяснить только повышенной активностью нейрогормонов.
Так, если главная причина и движущая сила ХСН - это влияние избытка
нейрогормонов, то блокада их действия (например, с помощью ингибиторов
ангиотензинпревращающего фермента (ИАПФ) по идее должна приводить
к 100% положительному клиническому результату. В реальной жизни
ИАПФ позволяют добиться снижения риска смерти больных ХСН в среднем
лишь на 23% [1]. Или другой пример. Как известно из результатов
исследования SOLVD [2], постоянное 4-летнее применение эналаприла
у больных с дисфункцией левого желудочка (ЛЖ) и клиническими признаками
сердечной недостаточности приводило к снижению риска смерти в
среднем на 16%. Однако, как видно из рисунка, достоверное влияние
препарата проявлялось лишь в первые 18 мес терапии, после чего
оставшиеся 30 мес кривые выживаемости больных в группе активного
лечения и плацебо шли абсолютно параллельно. Эти факты - отсутствие
100% клинического эффекта нейрогуморальных модуляторов и постепенное
исчезновение эффекта ИАПФ после нескольких месяцев терапии - могут
иметь несколько объяснений. Наиболее популярные из них обычно
связывают со следующим [3]:
Во-первых, ни один из существующих нейрогуморальных
модуляторов не оказывает полного
блокирующего влияния на гормональные системы, причастные к возникновению
и прогрессированию сердечной недостаточности;
Во-вторых, нейрогуморальные модуляторы теряют
свой эффект при более или менее длительном применении (так называемый феномен ускользания синтеза альдостерона
при применении ИАПФ и развитие "привыкания" при применении бета-блокаторов).
Другим реальным объяснением этих фактов может
явиться то, что наряду с нейрогормонами центральную роль в патогенезе
заболевания играют еще какие-то другие (в частности, иммунные)
механизмы, причастность которых к ХСН и обусловливает "неполную
компетентность" нейрогуморальной теории.
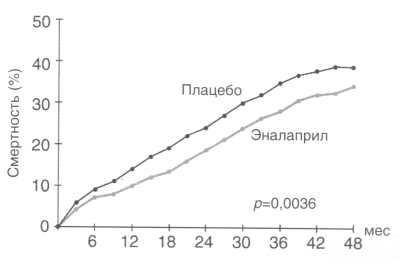
Рисунок. Исследование SOLVD.
Кривые смертности в
группе плацебо и эналаприла.С 18-го месяца лечения кривые "движутся"
параллельно.
Иммунная система и ХСН
Причастность иммунной системы к патогенезу
ХСН лишь на первый взгляд может показаться странной: хорошо известно,
что иммунная защита организма "срабатывает" не только при инфекционных
агрессиях, но реагирует также на любое стрессовое воздействие,
в том числе ишемию, гемодинамическую перегрузку, интоксикацию
и т.д., т.е. на те воздействия, которые являются причиной развития
и сердечной недостаточности. Существует несколько взаимосвязанных
компонентов иммунной системы, которые могут быть задействованы
в патогенезе ХСН, и главные из них - провоспалительные цитокины,
молекулы адгезии, аутоантитела, оксид азота и эндотелин-1. Все
большее внимание в иммунологических исследованиях при сердечной
недостаточности в последнее время
уделяется хемокинам (белкам, индуцирующим миграцию моноцитов в
миокарде), а также иным молекулярным образованиям (неоптерин,
шоковые белки, компоненты оксидативного стресса), роль которых
в процессе формирования синдрома ХСН пока окончательно не определена.
Провоспалительные цитокины являются наиболее
важным и хорошо изученным классом биологически активных веществ,
оказывающих иммунное и/или воспалительное действие и имеющих отношение
к сердечной недостаточности. Подробная характеристика и детальное
описание функций этих веществ представлены в обзоре, данном в
этом номере журнала. Можно лишь напомнить, что к основным провоспалительным
цитокинам относятся фактор некроза опухоли a (ФНО-a), интерлейкин 1 (ИЛ-1) и ИЛ-6. Наиболее "важный" для развития
ХСН цитокин - фактор некроза опухоли ФНО-a был открыт в сыворотке больных
со злокачественными новообразованиями еще в 1975 г. Carswell и
соавт. [4] как низкомолекулярное белковое вещество, обусловливающее
распад опухоли. Десятилетием позже было доказано участие этого
медиатора в молекулярных механизмах развития кахексии, в том числе
и при сердечных заболеваниях. Однако прямая связь ФНО-a
с синдромом сердечной недостаточности была установлена лишь 10
лет назад: в 1990 г. Levine и соавт. [5] впервые показали, что
уровень ФНО-a в сыворотке больных с тяжелой
сердечной недостаточностью (III-IV функциональный класс NYHA)
на порядок выше, чем у здоровых лиц: 115+25 U/ml против 9+3 U/ml
соответственно. Причем повышение активности ФНО-a
было более выраженным (> 39 U/ml) у пациентов с более тяжелыми
клиническими проявлениями декомпенсации, большей степенью кахексии
(массой тела 82% от идеального) и повышенной активностью РААС.
В последующих работах неоднократно подтверждалась тесная корреляционная
связь уровня ФНО-a, ИЛ-1b и ИЛ-6 с тяжестью клинических
проявлений [6-8] и активностью нейрогуморального фона больных
ХСН [9, 10].
Механизм реализации гемодинамического и клинического
влияния провоспалительных цитокинов при сердечной недостаточности
является предметом специальных исследований. На сегодняшний день
очевидно, что это влияние складывается по крайней мере из четырех
ключевых составляющих [11]: 1) отрицательного инотропного действия;
2) ремоделирования сердца (необратимая дилатация полостей и гипертрофия
кардиомиоцитов (КМЦ); 3) нарушения эндотелий-зависимой дилатации
артериол; 4) усиления процесса апоптоза КМЦ и клеток периферической
мускулатуры.
Логично предположить, что отрицательное инотропное
действие цитокинов может лежать в основе таких характерных гемодинамических
признаков ХСН, как низкий сердечный выброс и высокое внутрисердечное
давление, а в сочетании с нарушением регуляции тонуса периферических
артериол - быть причиной гипотонии, свойственной поздним стадиям
сердечной недостаточности. Утрата важного компенсаторного механизма,
каким является эндотелий-зависимая релаксация артериол [12] в
периферической мускулатуре, может обусловливать появление таких
клинических симптомов ХСН, как снижение толерантности к физическим
нагрузкам и уменьшение силы и выносливости скелетной мускулатуры.
Но, пожалуй, наиболее важными для формирования синдрома ХСН являются
"долговременные" эффекты провоспалительных цитокинов, проявляющиеся
постепенным разрушением внеклеточного коллагенового матрикса миокарда,
дилатацией желудочков и гипертрофией КМЦ. Как было показано в
ряде исследований, данные изменения, лежащие в основе феномена
ремоделирования сердца, носят необратимый характер [13] и наряду
с цитокин-индуцированным усилением апоптоза кардиомиоцитов способствуют
возникновению и прогрессированию ХСН и ухудшению прогноза этих
больных. Существуют данные, свидетельствующие о том, что высокая
концентрация растворимой формы рецептора ФНО-a (pФНО-a-P) является наиболее независимым предиктором неблагоприятного
прогноза больных ХСН, превосходящим по своей точности и специфичности
все другие прогностические маркеры, даже такие признанные, как
фракция выброса, функциональный класс ХСН и потребление кислорода
на максимуме нагрузки (VO2max) [14].
Сердечная недостаточность и иммунный
ответ: что первично?
Факт патогенетической взаимосвязи сердечной
недостаточности и повышенной экспрессии цитокинов в настоящее
время уже ни у кого не вызывает сомнений. Основная дискуссия ведется
вокруг вопроса о причинно-следственном характере этой связи.
Усиление застоя и нарастающая гипоксия периферических
тканей и самого миокарда, свойственные сердечной недостаточности,
вполне могут стать первопричиной активации иммунной системы и
приводить к росту ФНО-a и других провоспалительных цитокинов. Такая "последовательность"
событий косвенно подтверждается прямопропорциональной зависимостью
уровня ФНО-a от тяжести ХСН: чем выше функциональный класс ХСН, тем
более выражена реакция иммунной системы и выше уровень цитокинов.
И наоборот: уменьшение степени гипоксии понижает активность иммунного
ответа [15].
Однако большинство исследователей отводят экспрессии
провоспалительных цитокинов не подчиненную роль, а ставят ее в
ряд первопричин развития и прогрессирования ХСН. Косвенным подтверждением
этой теории являются положительные результаты применения препаратов,
способных снижать синтез цитокинов и улучшать при этом клиническое
течение ХСН. Прямые же доказательства ведущей роли цитокинов в
патогенезе ХСН получены в классической экспериментальной работе
Bozkurt и соавт., в которой длительная инфузия ФНО-a
приводит не только к снижению сократимости миокарда, но и к необратимой
дилатации желудочков сердца крыс [13]. Причем существуют данные,
что умеренная экспрессия ФНО-a
в миокарде мышей и человека, сочетающаяся с классическими клиническими
и морфологическими признаками декомпенсированной кардиомиопатией
(ДКМП), сопровождается минимальными воспалительными изменениями
сердечной мышцы [16, 17]. Последний факт подчеркивает независимую
(от воспаления) роль цитокинов в патогенезе ХСН, но вызывает вопрос
об их источнике у больных с недостаточностью кровообращения.
Как и почему у больных ХСН повышается
уровень провоспалительных цитокинов?
Основная причина активации иммунитета у
больных ХСН при отсутствии воспаления остается не ясной. Существуют
три гипотезы, объясняющие причины и механизм повышения уровня
цитокинов (и ФНО-a
как ведущего из них) при сердечной недостаточности.
Самой популярной является гипотеза миокардиальной
продукции цитокинов. Известно, что здоровое сердце не "производит"
цитокинов. Однако экспериментальные исследования показывают, что
КМЦ способны продуцировать ФНО-a, причем количество "производимого"
цитокина находится в прямой зависимости от степени напряжения
стенки миокарда ("диастолического стресса") и тем больше, чем
выше уровень конечно-диастолического давления в левом желудочке
[18]. Эта закономерность касается и других биологически активных
веществ, таких как фактор роста или шоковые белки. Гипотеза миокардиальной
продукции цитокинов объясняет терапевтическую эффективность препаратов,
способных уменьшать диастолическое напряжение миокарда у больных
ХСН: сердечных гликозидов, диуретиков, вазодилататоров, ИАПФ.
Однако эта гипотеза не объясняет эффективности тех средств, которые
с успехом применяются при ХСН, но не обладают свойством устранять
диастолический стресс, например, бета-адреноблокаторов. Кроме
того, трудно представить, что количество одних только "миокардиальных"
цитокинов будет достаточно для системных изменений в периферических
тканях и мускулатуре (потеря массы, похудание), характерных для
поздних стадий ХСН. Поэтому согласно другой гипотезе важнейший
источник провоспалительных цитокинов при сердечной недостаточности
- периферические ткани и скелетная мускулатура.
Экстрамиокардиальная продукция цитокинов
стимулируется тканевой гипоксией и избытком свободных радикалов,
возникающим вслед за повреждением миокарда и падением сердечного
выброса [19, 20]. Избыток цитокинов в свою очередь нарушает механизм
эндотелий-зависимой релаксации периферических сосудов [11] и в
еще большей степени способствует усилению тканевой гипоксии и
нарушению окислительных процессов: так замыкается очередной "порочный"
круг патогенеза ХСН. Согласно третьей гипотезе причиной повышения
уровня цитокинов у больных ХСН являются бактериальные эндотоксины,
проникновение которых в организм осуществляется через отечную
стенку кишечника. Венозный застой в кишечнике, неизбежный при
повреждении миокарда и падении сердечного выброса, способствует
повышению проницаемости стенки для бактерий и/или их токсинов,
которые, проникая в кровоток и взаимодействуя с CD14-рецептором
иммунокомпетентных клеток, запускают синтез ФНО-a
и других цитокинов [21, 22]. Эта оригинальная гипотеза, впервые
высказанная Anker и соавт. [21], имеет немало веских доказательств.
Так, например, было показано, что моноциты больных ХСН (!), как
правило, демонстрируют повышенную чувствительность к липополисахаридам,
входящим в состав клеточной мембраны бактерий [23]. У больных
ХСН концентрация эндотоксина в плазме тем выше, чем более выражен
отек кишечной стенки [22], причем применение диуретиков снижает
уровень как эндотоксина, так и ФНО-a [22]. "Кишечное" происхождение эндотоксина у больных ХСН
подтверждается тем фактом, что его концентрация в печеночных венах
достоверно выше, чем в левом желудочке или легочных венах [24].
Тем не менее в рамки этой эндотоксиновой гипотезы не укладывается
тот факт, что повышение уровня цитокинов отмечается у больных
ХСН уже на ранних стадиях заболевания, когда застойные явления
на периферии (в кишечнике) еще не так выражены [7]. Повышение
уровня провоспалительных цитокинов практически отсутствует у больных
с сердечной недостаточностью, развившейся на почве легочной гипертензии,
констриктивного перикардита или диастолической дисфункции [25].
Таким образом, ни одна из гипотез не отвечает
полностью на все вопросы, связанные с причиной и механизмом повышения
уровня цитокинов при сердечной недостаточности. Вероятно, что
повреждение миокарда с последующей дилатацией полостей и ростом
напряжения стенок сердца в сочетании с гипоксией периферических
тканей и неизбежным при этом застоем в кишечнике приводят к активации
всех основных источников цитокинов - КМЦ, скелетной мускулатуры
и иммунокомпетентных клеток. Результатом этого является критическое
повышение уровня циркулирующих цитокинов, негативные сердечно-сосудистые
эффекты которых способствуют еще большему повреждению миокарда.
Так замыкается патологический "цитокиновый" круг патогенеза сердечной
недостаточности.
Терапия
"Цитокиновая" модель патогенеза ХСН предусматривает
возможность эффективного воздействия на течение заболевания с
помощью новых классов лекарственных препаратов - ингибиторов синтеза
ФНО-a
(веснаринона, пентоксифиллина) или ингибиторов активности ФНО-a (энтерасепт), первые клинические
испытания которых дали обнадеживающие результаты. Частично обзор
этих исследований представлен в статье, опубликованной в этом
номере журнала.
Тем не менее важно подчеркнуть, что и классические
средства лечения ХСН, к которым в первую очередь относятся ИАПФ,
своему успеху в значительной степени могут быть обязаны способности
положительно воздействовать на иммунную систему организма. К чести
нашего института, следует отметить, что его сотрудники М.Ю.Самсонов,
Е.Л.Насонов стали одними из первых в мире, кто еще в 1993 г. на
культуре клеток моноцитов показал иммуномодулирующий эффект ИАПФ
каптоприла [26]. Это принципиально важный вывод, который позволяет
утверждать, что ИАПФ способны снижать уровень ФНО-a не только благодаря гемодинамической разгрузке миокарда
и снижению диастолического стресса, но и вследствие подавления
препаратом синтеза провоспалительных цитокинов как в КМЦ, так
и в других источниках. Подтверждение этому было получено и в ряде
клинических исследований. Так, Liu и соавт. выявили достоверное
снижение уровня ФНО-a у больных ХСН на фоне терапии
четырьмя различными ИАПФ - периндоприлом, беназеприлом, эналаприлом
и фозиноприлом [27]. Аналогичные данные были получены при применении
и других ИАПФ [28], что свидетельствует о системном характере
антицитокинового действия этого класса препаратов. Антицитокиновый
эффект ИАПФ скорее всего опосредован снижением синтеза AII - нейрогормона,
стимулирующего выработку ФНО-a [29]. Таким образом, высокая
эффективность ИАПФ у больных ХСН может быть обусловлена не только
модулирующим нейрогуморальным, но и частично противовоспалительным
влиянием.
Возможность опосредованного влияния на иммунную
систему через воздействие на нейрогуморальные механизмы и ангиотензин
II, в частности, подтверждается результатами исследований с препаратами
класса блокаторов АТ1-рецепторов. В работе Tsutamoto и соавт.
применение кандесартана у больных ХСН приводило к достоверному
снижению уровня цитокинов ФНО-a, ИЛ-6, а также растворимых молекул
адгезии ICAM-1 и VCAM-1 [30]. Среди возможных механизмов этого
эффекта обсуждается не только блокада АТ1-рецепторов, но и компенсаторная
стимуляция АТ2-рецепторов КМЦ, которые могут быть ответственны
за экспрессию цитокинов.
Отчетливая связь нейрогормонов с медиаторами
воспаления прослеживается также на примере катехоламинов: существует
доказательство усиления экспрессии ФНО-a
при увеличении уровня норадреналина [29]. Этот факт может
служить еще одним объяснением эффективного использования бета-адреноблокаторов
в лечении больных ХСН: устранение избытка симпатического влияния
и подавление негативного действия цитокинов.
Следует добавить, что антицитокиновый эффект
хотя бы частично присутствует также у сердечных гликозидов [31],
диуретиков [22], антагонистов
Ca2+ [32] и даже
у некоторых антиаритмиков, в частности у амиодарона [33], т.е.
у всех основных средств терапии ХСН.
Заключение
Таким образом, "цитокиновая" модель патогенеза
не противоречит нейрогуморальной теории, а дополняет наши представления
о механизмах развития ХСН. Участие медиаторов воспаления в схеме
заболевания расширяет "базу терапевтического вмешательства" и
открывает новые перспективы для повышения эффективности лечения
декомпенсированных больных. Уже сейчас серьезно обсуждаются пути
воздействия на цитокиновое звено сердечной недостаточности от
стерилизации кишечника с помощью антибиотиков до блокады синтеза
цитокинов или рецепторов к ФНО-a. И не исключено, что вскоре
антицитокиновые препараты станут таким же обычным средством лечения
больных ХСН, как сердечные гликозиды или ИАПФ.
Литература:
1. Carg R., Yusuf S. Overview of randomized
trials of angiotensin-converting enzyme inhibitors on mortality
and morbidity in patients with heart failure. JAMA 1995; 273:
1450-6.
2. The SOLVD Investigators. Effect of enalapril
on survival in patients with reduced left ventricular ejection
fraction and congestive heart failure. N Engl J Med 1991; 325:
293-302.
3. А.А.Скворцов, С.М.Челмакина, Н.И.Пожарская,
В.Ю.Мареев. Модулирование активности системы нейрогуморальной
регуляции при хронической сердечной недостаточности. Рус. мед.
журн. 2000; 8 (2): 87-93.
4. Carswell E.A., Old L.J., Kassel R.L. et al.
An endotoxin-indused serum factor that causes necrosis of tumor.
Proc Natl Acad Sci USA 1975; 72: 3666-70.
5. Levine B., Kalman J., Mayer L. et al. Elevated
circulating levels of tumor necrosis factor in severe chronic
heart failure. N Engl J Med 1990; 323: 236-41.
6. Teasta M., Yeh M., Lee P. et al. Circulating
levels of cytokines and their endogenous modulators in patients
with mild to severe congestive heart failure due to coronary artery
disease or hypertension. J Am Coll Cardiol 1996; 28: 964-71.
7. Torre-Amione G., Kapadia S., Benedict C.
et al. Proinflammatory cytokine levels in patients with depressed
left ventricular ejection fraction: a report from of SOLVD. J
Am Coll Cardiol 1996; 27: 1201-6.
8. Ferrari R., Bachetti T., Confortini R. et
al. Tumor necrosis factor soluble receptors in patients with various
degrees of congestive heart failure. Circulation 1995; 92: 1479-86.
9. MacGowan G., Mann D.L., Kormos R.L. et al.
Circulating interleukin-6 in severe congestive heart failure.
Am J Cardiol 1997; 79: 1128-31.
10. Rauchhaus M., Koloczek V., Florea V.
et al. The relationship between tumor necrosis factor-a and natriuretic
peptides in patients with chronic heart failure. Eur J Heart Failure
1999; 1 (Suppl): 203.
11. Е.Л.Насонов, М.Ю.Самсонов, Ю.Н.Беленков,
Д.Фукс. Иммунопатология застойной сердечной недостаточности: роль
цитокинов. Кардиология 1999; 3: 66-73.
12. Vanderheyden M., Kersschot E., Paulus W.
Pro-inflammatory cytokines and endothelium-dependent vasodilatation
in the forearm. Eur Heart J 1998; 19: 747-52.
13. Bozkurt B., Kribbs S.B.,
Clubb F.J. et al. Pathophysiologically relevant concentrations
of tumor necrosis factor-a promote progressive left ventricular
dysfunction and remodeling in rats. Circulation 1998; 97: 1382-91.
14. Rauchhaus M., Dohner W., Koloczek V. et
al. Systemically measured cytokines are independently predictive
for increased mortality in patients with chronic heart failure.
J Am Coll Cardiol 2000; 35 (Suppl. A): 1183.
15. Hasper D., Hummel L., Kleber F.X. et al.
Systemic inflammation in patients with heart failure. Eur Heart
J 1998; 19: 761-5.
16. Habib F.M., Springall D.R., Davies G.J.
et al. Tumor necrosis factor and inducible nitric oxide synthase
in dilated cardiomyopathy. Lancet 1996; 347: 1151-5.
17. Torre-Amione G., Kapadia S., Lee J. et al.
Expression and functional significance of tumor necrosis factor
receptors in human myocardium. Circulation 1995; 92: 1487-93.
18. Kapadia S.R., Oral H., Lee J. et al. Hemodynamic
regulation of tumor necrosis factor-a gene and protein expression
in adult feline myocardium. Circ Res 1997; 81: 187-95.
19. Adams V., Jiang H., Yu J. et al. Apoptosis
in skeletal myocytes of patients with chronic heart failure is
associated with exercise intolerance. J Am Coll Cardiol 1999;
33: 959-65.
20. Keith M., Geranmayegan A., Sole M. et al.
Increased oxidative stress in patients with congestive heart failure.
J Am Coll Cardiol 1998; 31: 1352-6.
21. Anker S.D., Egerer K., Volk H-D. et al.
Elevated soluble CD 14 receptors and altered cytokines in chronic
heart failure. Am J Cardiol 1997; 79: 1426-30.
22. Neibauer J., Volk H-D., Kemp M. et al. Endotoxin
and immune activation in heart failure: a prospective cohort study.
Lancet 1999; 353: 1838-42.
23. Moore K., O'Garra A., de Wall Malefyt R.
et al. Interleukin-10. Ann Rev Immunol 1993; 11: 165-71.
24. Peschel T., Anker S.D., Ziegenbalg K. et
al. Endotoxemia in congestive heart failure: highest levels in
hepatic veins suggestive of intestinal bacterial and/or endotoxin
translocation. Eur J Heart failure 2000; 2 (Suppl. 2): P22/10452.
25. Gurlek A., Kilikcap M., Dandachi R. et al.
Tumor necrosis factor-alpha in diastolic heart failure. Eur J
Heart failure 2000; 2 (Suppl. 2): P28/10381.
26. Samsonov M., Nassonov E., Werner-Felmayer
G. et al. Captopril and the effect of interferon gamma on monocytes.
Arch Intern Med 1993; 153: 1138-42.
27. Liu L., Zhao S-P. The changes of circulating
tumor necrosis factor levels in patients with congestive heart
failure influenced by therapy. Intern J Cardiol 1999; 69: 77-82.
28. Fukuzawa M., Satoh J., Sagara M. et al.
Angiotensin converting enzyme inhibitors suppress production of
tumor necrosis factor-alpha in vitro and in vivo. Immunopharmacology
1997; 36 (1): 49-66.
29. Koller-Strametz J., Pacher R., Fery B. et
al. Circulating tumor necrosis factor levels in chronic heart
failure: relation to its soluble receptor II, interleukin-6 and
neurohumoral variables. J Heart Lung transplant 1998; 17: 356-62.
30. Tsutamoto T., Wada A., Maeda K. et al. Angiotensin
II type receptor antagonist decreases plasma levels of tumor necrosis
factor alpha, interleukin-6 and soluble adhesion molecules in
patients with chronic heart failure. J Am Coll Cardiol 2000; 35:
714-21.
31. Matsumori A., Ono K., Nishio R. et al. Modulation
of cytokine production and protection against lethal endotoxemia
by the cardiac glycoside oubaine. Circulation 1997; 96: 1501-6.
32. Mohler E.R., Sorensen L.C., Ghall J.K. et
al. Role of cytokines in the mechanism of action of amlodipine:
the PRAISE heart failure trial. J Am Coll Cardiol 1997; 30: 35-41.
33. Matsumori A., Ono K., Nishio R. et al. Amiodarone
inhibits production of tumor necrosis factor alpha by human mononuclear
cells. Circulation 1997; 96: 1386-9.