Терапия-Дилатационная кардиомиопатия сегодня
Дилатационная кардиомиопатия сегодня
С.Н.Терещенко, Н.А.Джаиани
Кафедра внутренних болезней РУДН, Москва
URL
P>Список сокращений:
| БАБ - бета-адреноблокаторы | ИАПФ - ингибиторы ангиотензинпревращающего фермента |
| ДКМП - дилатационная кардиомиопатия | ХСН - хроническая сердечная недостаточность |
Впервые термин "кардиомиопатия" был предложен W.Bridgen в
1957 г. Согласно его определению кардиомиопатия – это группа болезней миокарда
неизвестной этиологии некоронарогенного происхождения. На протяжении длительного
времени это понятие неоднократно изменялось, порождая путаницу в терминологии. В
последствие, благодаря внедрению современных методов диагностики, как
инвазивных, так и неинвазивных, удалось установить происхождение многих
кардиомиопатий, и Всемирной Организацией здоровья предложены классификации,
последняя из которых представлена в 1995 г. [1] и делит кардиомиопатии на:
1. Дилатационную.
2.
Гипертрофическую.
3. Рестриктивную.
4.
Специфическую (метаболические, воспалительные, ишемические, клапанные и др.). К
метаболическим относятся диабетическая, алкогольная кардиомиопатия и
прочие.
5. Аритмогенную кардиомиопатию правого
желудочка.
6. Неклассифицируемые кардиомиопатии
(фиброэластоз и др.).
Таким образом, кардиомиопатии – это
неоднородная группа хронических заболеваний в большинстве случаев неизвестной
этиологии, за исключением специфических. Специфические кардиомиопатии по
структурно-функциональному состоянию миокарда ближе к дилатационной. Однако они
не соответствуют определению дилатационной кардиомиопатии. В связи с этим не
утихают споры в отношении того, имеют ли право на существование ишемическая,
диабетическая кардиомиопатии и др. В настоящее время в зарубежной литературе
часто встречаются эти термины. На наш взгляд использование этих терминов
необходимо, так как это упрощает понимание тяжести состояния больного, у
конкретного больного имеется выраженная дилатация с резким нарушением
сократительной функции левого желудочка. Однако нельзя в этих ситуациях
использовать термин "дилатационная кардиомиопатия" (ДКМП).
ДКМП являясь самой распространенной, встречается во всех
странах мира. Имеющиеся до недавнего времени разногласия по вопросам определения
кардиомиопатий и отсутствие четких диагностических критериев ДКМП обусловливают
трудности проведения эпидемиологических исследований в этой области, в связи с
чем на сегодняшний день точные данные о распространенности ДКМП и заболеваемости
населения отсутствуют, поскольку большинство исследований носят ретроспективный
характер и основываются на анализе лишь точно установленных диагнозов без учета
ранних стадий болезни. По результатам таких исследований можно приблизительно
судить о частоте возникновения ДКМП. Удельный вес ДКМП среди других
кардиомиопатий составляет 60%. В этом отношении не потеряло своего значения
высказывание Н.М.Мухарлямова: "Необходимы серьезные эпидемиологические
исследования, которые позволят выяснить истинное состояние дел. Важность этой
проблемы подчеркивается и тем, что больные ДКМП быстрее, чем при других
некоронарогенных заболеваниях миокарда, становятся стойкими инвалидами" [2].
Патогенез ДКМП
В
настоящее время большой интерес представляют ДКМП не установленной этиологии,
так называемые идиопатические ДКМП. Многочисленные исследования последнего
десятилетия ориентируются на изучение их этиопатогенеза, и в этом аспекте
рассматриваются гипотезы хронической вирусной инфекции, аутоиммунного влияния и
генетической детерминированности [2–8]. Стали доступны молекулярно-биологические
технологии (в том числе, полимеразная цепная реакция), с помощью которых
выявлена роль энтеровирусов, в частности, группы В коксакивирусов
[2, 5, 9, 10], в патогенезе ДКМП. Несмотря на высокую чувствительность и
специфичность этих технологий, частота выявления вирусов варьирует от 0 до 40%
[8]. У детей в возрасте от 1 дня до 19 лет с быстроразвивающейся дилатацией
левого желудочка и его дисфункцией вирусный геном выявлялся в 68% случаев,
причем энтеровирус встречался в 30% случаев, аденовирус – в 58%, герпесвирус –
8%, цитомегаловирус - в 4% [8].
Аутоиммунное
влияние на развитие идиопатической ДКМП изучено больше на гуморальном
иммунитете. Имеются сообщения о наличии кардиальных органоспецифических
аутоантител [11], таких как антимиозин, антиактин, антимиолемма,
анти-альфа-миозин и анти-бета-миозин тяжелых цепей, последние две
характеризуются высокой специфичностью для кардиомиоцитов и вставочных дисков.
Выявлен также анти-аденозин-дифосфат-аденозин-трифосфат, представляющий антитела
к митохондриальной мембране кардиомиоцита [8] и оказывающий неблагоприятное
влияние на функционирование мембранных кальциевых каналов, что в свою очередь
приводит к нарушению метаболизма миокарда.
Однако подобные открытия являются лишь следствием причинного фактора, который
еще необходимо установить.
Следует отметить, что
кардиоспецифические антитела в большинстве случаев выявлялись при семейных ДКМП,
следовательно генетические факторы могут иметь большое значение в развитии
идиопатической ДКМП, что стало очевидно в результате многих
работ.
Важнейшим открытием для медицины стала возможность
молекулярной генетики идентифицировать гены, отвечающие за развитие тех или иных
заболеваний. С этой точки зрения большие успехи достигнуты при изучении
генетического базиса идиопатических ДКМП. Примерно треть случаев
идиопатических ДКМП определены как семейные [12], при которых преимущественно
превалирует аутосомно-доминантное наследование (аутосомно-доминантные ДКМП).
Наряду с аутосомно-доминантными описываются аутосомно-рецессивные, Х-сцепленные,
митохондриальные ДКМП [13].
Аутосомно-доминантные формы
характеризуются клинической вариабельностью и генетической гетерогенностью. Эти
формы ассоциируются с шестью различными локусами [13, 14]: так называемая
простая ДКМП - с локусами 1q32,2p31,9q13,10q21–q23, тогда как ДКМП с нарушениями
проводимости – с локусами 1q1–1q1, 3p22–3p25, причем неизвестно, за синтез каких
кардиальных белков отвечают эти локусы.
Установлено, что мутации кардиального актина локализуются в локусах 9q13–22 и
1q32 [13], а также в локусе 15q14 [15].
Митохондриальные ДКМП являются следствием
аномалии митохондриальной структуры и дисфункции процесса окислительного
фосфорилирования [16]. Как известно, митохондрии имеют собственную ДНК,
содержащую всего лишь 37 генов, и свои механизмы транскрипции и трансляции.
Митохондриальные ДНК отличаются от геномных ДНК тем, что первые не имеют
интронов, защитных гистонов, эффективных ДНК-восстановительных систем,
следовательно частота мутаций митохондриальных ДНК в 10 раз выше, чем в ядерных
геномных ДНК [17]. В каждой митохондрии имеется одиночная хромосома, кодирующая
ряд ферментов (13 из 69), участвующих в механизме окислительного
фосфорилирования. Следовательно, вследствие мутации нарушается энергетический
обмен кардиомиоцитов, что ведет к развитию ДКМП.
| От редакции |
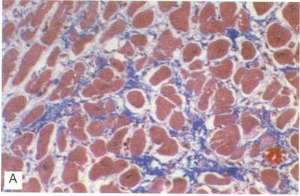

Рис. Дилатационная кардиомиопатия. А. Гистологический препарат миокарда при ДКМП. Отмечается гипертрофия отдельных кардиомиоцитов и интерстициальный фиброз (коллаген окрашен в синий цвет по Masson). В. Макропрепарат сердца при ДКМП. Имеет место дилатация всех полостей сердца с одновременной гипертрофией миокарда (масса сердца увеличена в 3 раза). Обращает на себя внимание хорошее состояние коронарных артерий. В области верхушки левого желудочка имеются небольшие пристеночные тромбы (которые при ДКМП становятся источником тромбоэмболических осложнений).
Описаны точечные мутации и
множественные делеции в митохондриальных ДНК как при спорадических случаях ДКМП,
так и при семейных. Многие митохондриальные миопатии ассоциируются с
неврологическими нарушениями: митохондриальные ДКМП встречаются при таких
митохондриальных синдромах, как синдром HELAS (митохондриальная миопатия,
энцефалопатия, лактацидоз, эпизоды нарушения мозгового кровообращения), MERRF
(сопровождается миоклонус-эпилепсией), Kearus-Sayre-синдром (KSS), при синдроме
дефицита НАДН- коэнзим Q редуктазы. При синдромах MELAS и MERRF выявлены
точечные мутации, делеции найдены при синдроме KSS [8,
17].
Существенный прогресс достигнут в изучении
молекулярных основ Х-сцепленных ДКМП. Описаны мутации различных
участков гена, отвечающего за синтез белка дистрофина (21 хромосома) [8, 17,
18]. Дистрофин – миокардиальный белок, входящий в состав мультипротеинного
комплекса, который связывает мышечный цитоскелет кардиомиоцита с внеклеточным
матриксом, благодаря этому происходит скрепление кардиомиоцитов в
экстрацеллюлярном матриксе. В клетке дистрофин связан непосредственно с
сократительным белком актином [19, 20]. Таким образом, дистрофин выполняет ряд
важнейших функций: 1) мембраностабилизирующую; 2) передает сократительную
энергию кардиомиоцита во внеклеточную среду; 3) обеспечивает мембранную
дифференциацию, т.е. специфичность мембраны кардиомиоцита [21]. Выявлены
мутации, при которых происходит замена нуклеотидов [8, 20], в результате чего
синтезируются аминокислоты, которые нарушают полярность и другие свойства
дистрофина как белка, поэтому теряется мембраностабилизирующее свойство
последнего. Итогом является дисфункция
кардиомиоцита.
Мутации гена дистрофина описаны также при
ДКМП, ассоциированных с мышечными дистрофиями Дюшена, Беккера, чаще всего в этих
случаях выявлялись делеции [8]. Однако популяционных исследований по выявлению
мутации гена дистрофина у неродственных больных идиопатической ДКМП не
проводилось. Нами было проведено исследование по выявлению мутации генов
кардиального актина и дистрофина у 20 больных идиопатической кардиомиопатией.
Несмотря на теоретические предпосылки и наши ожидания, мутации этих генов у
исследуемых нами больных выявлено не было. То, что в нашей работе не выявлялась
мутация генов актина и дистрофина, не является прямым утверждением того, что
мутации действительно нет при несемейной кардиопатии. Вероятно, для глубоких
выводов необходимо большое популяционное исследование. И, может, не менее
значимым было бы исследование генов других структурных компонентов сердечной
мышцы, в частности, коллагена и эластина, мутация которых, возможно, тоже имеет
значение в развитии ДКМП [22].
При мышечной дистрофии
Эмери-Дрейфуса (Х-сцепленной), одной из проявлений которой является ДКМП,
выявлена мутация гена, отвечающего за синтез белка эмерина (28-я хромосома).
Эмерин является компонентом оболочки ядра кардиомиоцита и скелетной мускулатуры,
поэтому наряду с ДКМП заболевание характеризуется также наличием суставных
контрактур. Дебютирует заболевание чаще в возрасте от 2 до 10 лет, когда
появляется слабость в мышцах плечевого пояса и верхних конечностей
[17].
Что касается несемейных случаев идиопатической
ДКМП, описано нарушение в экспрессии гена белка метавинкулина. Последний
является белком цитоскелета кардиомиоцита и связывает актин со вставочными
дисками. Исследованы 23 больных с идиопатической ДКМП, с помощью полимеразной
цепной реакции выявлено нарушение транскрипции метавинкулина и отсутствие этого
белка в тканях сердечной мышцы [23].
В ряде работ выявлена
усиленная экспрессия генов внеклеточных белков металлопротеиназ, одним из
представителей которых является интерстициальная коллагеназа; у больных с
идиопатической ДКМП отмечено 3–4-х кратное повышение уровня последнего в
сердечной ткани [24].
Проводилась связь между полиморфизмом
гена АПФ и идиопатической ДКМП. В трех работах (в двух из них соотношение
больных и здоровых составило 112/79, 81/40 соответственно, в другой – 99 больных
ДКМП, 364-контроль) отмечена корреляция DD-генотипа с ДКМП. В настоящее время во
Франции в этом плане проводится исследование 433 больных с идиопатической
ДКМП [18].
Таким образом, роль
генетических факторов бесспорна в этиопатогенезе идиопатических ДКМП.
Исследования в этой области необходимы для оценки генетического риска развития
заболевания. И это понятно, так как происходит рост заболеваемости и смертности
от данной патологии.
Лечение ДКМП
Наряду
с новшествами в патогенезе ДКМП, последнее десятилетие ознаменовано появлением
новых взглядов на его лечение. Как известно, важнейшим клиническим
проявлением ДКМП является хроническая сердечная недостаточность (ХСН).
Нужно отметить, что в клинической практике прогрессирующая сердечная
недостаточность оказывается часто дебютом ДКМП и, особенно, идиопатической формы
заболевания. Поэтому лечение ХСН является важным моментом в ведении больных с
ДКМП любой этиологии. Современная терапия направлена не только на устранение
симптомов сердечной недостаточности, но и на предотвращение возникновения и
прогрессирования ХСН. В связи с этим большим достижением стало более тщательное
изучение свойств ингибиторов ангиотензинпревращающего фермента (АПФ). Последние
показали не только способность увеличивать фракцию выброса левого желудочка,
повышать толерантность больных к физической нагрузке и в некоторых случаях
улучшать функциональный класс недостаточности кровообращения, как это
продемонстрировали исследования начала 80-х годов [25], но и улучшение прогноза
жизни (CONSENSUS, SOLVD), снижение смертности, рост выживаемости у больных с
низкой фракцией выброса. Поэтому ингибиторы АПФ являются препаратами
первого ряда в лечении больных с ХСН. Назначение этих средств показано
на всех стадиях симптоматической сердечной недостаточности, связанной с
систолической дисфункцией миокарда [26].
Не менее значимым
обстоятельством последние годы стал пересмотр точки зрения на
бета-адреноблокаторы (БАБ). В 90-е годы в результате многоцентровых
плацебо-контролируемых исследований кардиологи пришли к единому утверждению о
возможности назначения этих отрицательных инотропных средств в лечении ХСН. БАБ,
воздействуя на гиперактивацию симпато-адреналовой системы, показали способность улучшать
гемодинамику и течение сердечной недостаточности, оказывать протективное
действие на кардиомиоциты, снижать тахикардию и, соответственно, ишемию
миокарда, предотвращать нарушения ритма [9, 27]. Исследование CIBIS выявило
снижение заболеваемости и частоты госпитализаций, а также смертности при
применении кардиоселективного БАБ бисопролола. Достоверно благоприятный эффект
при этом отмечен у больных с неишемической этиологией ХСН, в частности, с ДКМП,
у больных тяжелой декомпенсацией (IV функциональный класс по NYHA). Исследование
CIBIS-II доказало способность бисопролола снижать риск смерти больных, число
госпитализаций. Положительное действие в результате исследований (в том числе,
больных с ДКМП) выявлено также при применении некардиоселективного БАБ
карведилола, обладающего свойствами альфа-блокатора, вазодилататора и
антиоксиданта [9].
Таким образом, БАБ улучшают
прогноз и выживаемость больных с ХСН. Они рекомендованы для терапии при
этом в качестве основных препаратов. Другое дело, что использовать их
необходимо, учитывая противопоказания, с медленным титрованием дозы, начиная с
минимальных дозировок, и их следует применять дополнительно к терапии
ингибиторами АПФ, мочегонными и сердечными гликозидами (если последние
необходимы).
Последнее время все чаще при неудачах в
консервативной терапии ДКМП рассматривается вопрос о трансплантации сердца,
выживаемость при этом, по данным ряда авторов, составляет более 70% через 10 лет
[15]. Однако проблема пересадки сердца, наряду с высокой стоимостью
вмешательства, состоит и в недостатке донорского органа. В связи с этим в
настоящее время клиническую оценку проходят механические устройства обхода
желудочков [26, 28].
Литература
1.
Sinagra G, Mestroni L, Camerini F. The classification of cardiomyopathies.
Cardiomyopathies 1999; p.3–8.
2. Мухарлямов Н.М., Попович
М.И., Затушевский И.Ф. Дилатационная кардиомиопатия. Кишенев: "Штиинца", 1986;
158 с.
3. Амосова Е.Н. Кардиомиопатии. Киев: "Книга Плюс",
1999; 421 с.
4. Кушаковский М.С. Хроническая застойная
сердечная недостаточность. Идиопатическаие миокардиопатии. СПб: "Фолиант", 1998;
320 с.
5. Моисеев В.С., Сумароков А.В, Стяжкин В.Ю.
Кардиомиопатии. М.: Медицина 1993; 176 с.
6. Моисеев В.С.
Сердечная недостаточность и достижения генетики. // Сердечная недостаточность
2000; 4: 121–31.
7. Терещенко С.Н., Джаиани Н.А., Моисеев
В.С. Генетические аспекты хронической сердечной недостаточности//Тер. арх. 2000;
4: 75–7.
8. Mestroni L, Rocco C. et al. Advances in
molecular genetics of dilated cardiomyopathy//Cardiology Clinics 1998; 16:
603-9.
9. Мареев В.Ю. Бета-адреноблокаторы – новое
напрваление в лечении хронической сердечной недостаточности//Рус. мед. журн.
1999; 2: 76-–8.
10. Fujioka S,
Koide H, Kitaura Y. et al. Molecular detection and differentiation of
enteroviruses in endomyocardial biopsies and pericardial effusions from dilated
cardiomyopathy and myocarditis//Am Heart J 1996; 131: 760–5.
11. Caforio ALP, Crazzini M, Mann J.M., et al.
Identification of alfa and beta- cardiac myosin heavy chain isoforms as major
autoantigens in dilated cardiomyopathy//Circulation 1992; 85: 1734–42.
12. Fatkin D, MacRai C. et al. Missense mutations in the
rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and
conduction system disease//N Engl J Med 1999; 341: 1715-26.
13. Komajda M, Charron P, Tesson F. Genetic aspects of
heart failure//Eur J Heart Failure 1999; 121–6.
14. Priori
S, Barhanin J., et al. Genetic and molecular basis of cardiac arrhythmias//Eur
Heart J 1999; 20: 174–95.
15. Olson T, Michels V., et al.
Actin mutations in dilated cardiomyopathy, a heritable form of heart
failure//Sciense 1998; 280.
16. Bachinski L., Roberts R.
Causes of dilated cardiomyopathy//Cardiology clinics 1998; 16.
17. Towbin J, Bowle S K, Ortiz-Lopez R, Wang Q. Genetic
basis of dilated cardiomyopathy. Cardiomyopathies 1999; 56-65.
18. Dec G, Fuster V. Idiopathic dilated cardiomyopathy//N
Engl J Med 1994; 331: 1564-75.
19. Leiden J.M. The
genetics of dilated cardiomyopathy – emerging clues to the puzzle//New Engl J
Med 1997; 337: 1080–1.
20. Sakamoto A., Ono K., Abe M.,
Jasmin G., Eki T., Murakami Y., Masaki T., Toyooka T., Hanaoka F. Both
hypertrophic and dilated cardiomyopathies are caused by mutation of the same
gene, delta-sarcoglycan, in hamster: an animal model of disrupted
dystrophin-associated glycoprotein complex//Proc Natl Acad Sci USA 1997; 94:
13873–8.
21. Ortiz-Lopez R., Li H., Su J. et al. Evidens for a dystrophin
missense mutation as a cause of X-linked dilated cardiomyopathy//Circulatoin
1997; 95: 2434–40.
22. Терещенко С.Н., Джаиани Н.А.,
Мареев В.Ю. Влияние генов, отвечающих за синтез кардиальных белков актина и
дистрофина, на развитие хронической сердечной недостаточности у больных с
инфарктом миокарда и дилатационной кардиомиопатии//Сердечная недостаточность
2000; 1: 18-20.
23. Maeda M, Holder E, Lowes B., et al.
Dilated cardiomyopathy assotiated with deficiensy of
the cytoskeletal protein metavinculin. Circulation 1997; 95 (1):17–20.
24. Tyagi S, Kumar S, Voelker DJ, et al. Differential gene
expression of extracellular matrix components in dilated cardiomyopathy. J Cell
Biochem 1996, november 1; 63 (2): 185–98.
25. Сидоренко Б.А., Преображенский Д.В.
Лечение и профилактика сердечной недостаточности. М. 1997;
92–8.
26. Лечение сердечной недостаточности. Рекомендации
рабочей группы по изучению сердечной недостаточности Европейского Общества
Кардиологов. Рус. мед. журн. Приложение. 1999.
27. Терещенко
С.Н., Демидова И.В. Хроническая сердечная недостаточность: диагностика и
лечение. Методические рекомендации. М. 2000; 26 с.
28.
Gronda E., Vitali E. Left ventricle assist systems: a possible
alternative to heart transplantation for heart
failure patients? Patient selection, techniques and benefit. Eur J Heart Failure
Dec 1999; 1: 320–5.