Онкология-
Опухолевые супрессоры и мутаторные гены
Б.П.Копнин
Российский онкологический научный центр им. Н.Н.Блохина РАМН, Москва
источник RosOncoWeb.Ru
01 02 03 04 05 06 073.11. Мутаторные гены
Нарушения функций вышерассмотренных белков, контролирующих апоптоз и/или клеточный цикл (p53, pRb, p16INK4a, pARF и др.) отменяют запрет на пролиферацию клеток с различными аномалиями, в том числе и с генетическими изменениями, что увеличивает вероятность появления онкогенных клеточных клонов. Эту группу белков принято называть "gatekeepers" - "сторожи". Наряду с этим идентифицирован ряд компонентов специализированных систем распознавания и репарации повреждений ДНК, дисфункция которых также вызывает генетическую нестабильность, предопределяющую развитие новообразований. Они получили название "caretakers" - "смотрители". Эта вторая группа белков и является предметом рассмотрения данного раздела.
В зависимости от типа повреждений ДНК могут активироваться три типа репарационных систем: а) системы репарации двунитевых разрывов ДНК; б) системы репарации неспаренных оснований ("mismatch repair"); и, в) системы эксцизионной репарации. Описаны наследственные формы новообразований, связанные с врожденными мутациями генов, продукты которых обеспечивают активацию и функционирование каждой из этих систем. Причем, некоторые из этих белков (ATM, CHK2, р53, BRCA1) активируют также и молекулы, ответственные за остановку клеточного цикла и индукцию апоптоза, выполняя, таким образом, одновременно функции и "смотрителя", и "сторожа".
3.11.1. АТМ, ATR, NBS1, CHK1 и CHK2 - компоненты систем проведения сигналов от поврежденной ДНК к различным эффекторам
Ключевую роль в интеграции сигналов от поврежденной ДНК и их дальнейшей передаче к разнообразным эффекторам играют специфические протеинкиназы ATM (Ataxia-Telangiectasia Mutated), ATR (ATM Related), NBS1, CHK1 и CHK2 (чекпойнткиназы 1, 2) - Рис. 7. Белок ATM, имеющий структурное сходство с фосфатидилинозит-3-киназой (PI3K), накапливается в местах повреждений и приобретает киназную активность, связывая фосфорилированные белки хроматина (H2AX и др.) и белки-сенсоры нарушений структуры ДНК. Причем, ATM активируется в ответ на возникновение двунитевых разрывов ДНК (вызываются g-облучением, ингибиторами топоизомераз и т.д.), тогда как другие нарушения структуры ДНК (например, сшивки оснований, вызываемые УФ-облучением или повреждения, индуцируемые алкилирующими соединениями) не активирует ATM. В этих случаях, как и при ингибировании синтеза ДНК, наблюдается функциональная активация гомолога ATM, белка ATR. Активированные формы ATM и ATR фосфорилируют ряд своих мишеней, в частности р53 (см. раздел 3.3.3), Mre11, NBS1, CHK1, CHK2 и BRCA1 (Рис. 7).
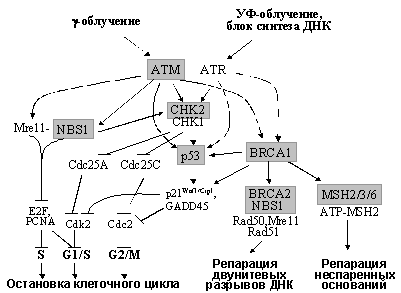
Рис. 7. Схема сигнальных путей, регулирующих реакции клетки на повреждения ДНК. Выделены компоненты, герминальные мутации которых ответственны за наследственные синдромы, характеризующиеся предрасположенностью к развитию определенных новообразований.
Причем для фосфорилирования CHK2 необходимо предварительное фосфорилирование белков комплекса Mre11/NBS1/Rad50, который, локализуясь в местах повреждений, рекрутирует к ним различные молекулы, в том числе CHK2, BRCA1, E2F и PCNA. Привлечение PCNA вызывает переключение с репликативного синтеза ДНК на репарационный и остановку клеточного цикла в S фазе; к блокированию входа и продвижения по S ведет и подавление функции E2F (см. 3.2.2). Фосфорилированные чекпойнткиназы CHK1/2, в свою очередь, фосфорилируют и инактивируют белки семейства Cdc25, что вызывает подавление активности регулируемых ими циклинзависимых киназ и быструю остановку клеточного цикла в G1 (если Cdc25A не активирует Cdk2) или в G2 (когда Cdc25C не активирует Cdc2). Кроме того, CHK1 и CHK2 амплифицируют сигналы к р53 и BRCA1, что способствует длительной задержке в G1 или G2 (см. разделы 3.3.3 и 3.11.2) и, кроме того, активизирует системы репарации ДНК (см. следующие разделы) - Рис.7.
Герминальные инактивирующие мутации обоих аллелей гена ATM вызывают атаксию-телангиэктазию (АТ) - тяжелое заболевание, характеризующееся нейродегенерацией, иммунодефицитом и повышенным риском возникновения новообразований. Примерно у 10% пациентов с АТ в молодом возрасте развиваются лимфоидные опухоли из Т- или В-клеток (лимфосаркомы, лимфогрануломатоз, различные формы лейкозов), а также рак молочной железы. Соматические гомозиготные мутации гена АТМ характерны и для некоторых форм ненаследственных лимфолейкозов (Т-клеточного пролимфоцитарного лейкоза, В-клеточного хронического лимфолейкоза и др.). Гомозиготный нокаут гена ATM у мышей также значительно увеличивает вероятность развития лимфоидных неоплазий. У индивидуумов с герминальными мутациями только одного из двух аллелей гена АТМ несколько повышена частота возникновения рака молочной железы. Онкогенный потенциал мутаций АТМ связан, очевидно, с нарушениями реакций клетки на повреждения ДНК и возникающей в связи с этим генетической нестабильностью. Так, после g-облучения в клетках с дефектным АТМ не происходит полноценной активации чекпойнтов и остановки клеточного цикла в G1, S или G2. Кроме того, в них блокирована активизация системы репарации двунитевых разрывов ДНК. В результате при инактивации АТМ резко увеличивается вероятность размножения клеточных вариантов с различными генетическими нарушениями.
Сходные последствия наблюдаются и при инактивации одной из важнейших мишеней АТМ - белка NBS1. Герминальные гомозиготные мутации гена NBS1 вызывают Ниймегенский синдром (Nijmegen Breakage Syndrome), характеризующийся иммунодефицитом, генетической нестабильностью и повышенной предрасположенностью к развитию лимфоидных новообразований (в отличие от мутаций АТМ, мутации NBS1 не вызывают атаксию и телангиэктазию). Соматические мутации гена NBS1 выявляются в 10-20% случаев ненаследственных форм острого лимфобластного лейкоза. В клетках с инактивацией NBS1 наблюдается отмена остановки в S после g-облучения и понижение эффективности работы систем репарации двунитевых разрывов ДНК вследствие нарушения функционирования комплекса Rad50/Mre11/NBS1, обеспечивающего оба механизма исправления таких повреждений - гомологичную рекомбинацию ДНК и воссоединение концов разорванной ДНК.
Потенциальным онкогенным эффектом обладают, по-видимому, и нарушения функции белка ATR. Гетерозиготный нокаут гена ATR у мышей приводит к увеличению частоты возникновения лимфосарком, фибросарком, раков печени и яичника (инактивация обоих аллелей гена ATR, в отличие от гомозиготного нокаута гена ATM, вызывает внутриутробную гибель). У людей наследственного предрасположения к развитию каких-либо новообразований, связанного с врожденными мутациями ATR пока не выявлено, но соматические мутации этого гена нередко выявляются в клетках некоторых опухолей, в частности рака желудка.
Увеличение риска развития новообразований наблюдается и при врожденных мутациях чекпойнткиназы CHK2. Оказалось, что у части пациентов с клиническими проявлениями синдрома Ли-Фраумени (см. раздел 3.3.1), но не имеющих мутаций р53, выявляются герминальные гетерозиготные мутации гена CHK2. Этот факт свидетельствует о ключевой роли нарушений сигнального пути CHK2-p53, контролирующего реакции клетки на повреждения ДНК, в возникновении сильной предрасположенности к развитию самых разных новообразований. Соматические инактивирующие мутации чекпойнткиназ CHK2 и CHK1 обнаруживаются в части случаев наиболее распространенных опухолей: рака легкого, толстой кишки, матки и др.
3.11.2. BRCA1 и BRCA2 контролируют репарацию ДНК и размножение клеток
Гены BRCA1 и BRCA2 были впервые идентифицированы как гены, врожденные мутации которых ассоциированы с наследственными формами рака молочной железы. У женщин с герминальными мутациями одного из аллелей гена BRCA1 риск развития в течение жизни рака молочной железы составляет около 85% (этот риск несколько варьирует в зависимости от местоположения и/или типа мутаций). Для опухолей яичника такой риск несколько меньше - около 50%. У носителей врожденных мутаций гена BRCA1 выше также вероятность развития опухолей толстого кишечника и простаты. При герминальных мутациях гена BRCA2 риск развития опухолей молочной железы несколько ниже, чем при мутациях BRCA1. Отличительными чертами мутаций BRCA2 являются более частое возникновение рака молочной железы у мужчин и меньший риск развития опухолей яичника. Гены BRCA1 и BRCA2 ведут себя как классические опухолевые супрессоры: для инициации опухолевого роста помимо врожденной мутации в одном из аллелей необходима и инактивация второго аллеля, которая происходит уже в соматической клетке. Как правило, мутации в генах BRCA1 и BRCA2 ведут к прекращению синтеза полноразмерного белка. Особенностью мутаций генов BRCA1 и BRCA2 является то, что они характерны для наследственных форм новообразований и значительно реже обнаруживаются в ненаследственных опухолях той же локализации.
Гены BRCA1 и BRCA2 кодируют ядерные фосфобелки (соответственно 1863 и 3495 аминокислот), которые за счет разнообразных белок-белковых взаимодействий участвуют в регуляции репарации ДНК и размножения клеток. Так, белок BRCA1 связывает белки, ответственные за гомологичную рекомбинацию и репарацию двунитевых разрывов ДНК (Rad50, Rad51, BRCA2), компоненты систем репарации неспаренных оснований ДНК (MSH2, MSH6, MLH1, ATP-MSH2 и др.), транскрипционные факторы (базальные - HDAC, р300/CBP, SWI/SNF; и сиквенс-специфические - p53, Myc, E2F, ZBRK1, ATF, рецептор эстрогенов, рецептор андрогенов), а также ряд других белков - pRb (см. II.3.2), BARD1 (опосредует убиквитинирование), BAP1 (ответственен за деубиквитинирование), Nm23 (компонент центросомы) и т.д.
Транскрипционная функция BRCA1 заключается в его способности репрессировать одни сиквенс-специфические факторы транскрипции (Myc, E2F, рецептор эстрогенов и др.) и активировать другие (р53 и др.) и модулировать таким образом активность генов, регулируемых этими факторами. При генотоксических стрессах (g-облучение и др.) транскрипционная функция BRCA1 направлена на индукцию остановки клеточного цикла по нескольких механизмам. Так, она обеспечивает усиление активности р53; включение дублирующих, р53-независимых, путей активации некоторых р53-респонсивных генов (p21Waf1/Cip1, GADD45), вызывающих задержку соответственно в G1 и G2 (см. раздел 3.3.3); подавление активности Myc, E2F и т.д. Одновременно активированный BRCA1, взаимодействуя с белками репарационных систем, стимулирует восстановление нормальной структуры ДНК. Рекрутируя комплексы Rad50/Mre11/NBS1, он стимулирует процессирование концов разорванной ДНК, подготавливая их либо для гомологичной рекомбинации, либо для воссоединения "конец в конец" - двух основных путей репарации двунитевых разрывов ДНК. Взаимодействуя с комплексом Rad51/BRCA2, он увеличивает эффективность процесса гомологичной рекомбинации ДНК. Связываясь с белками MSH2, MSH3, MSH6 и др., BRCA1 участвует, очевидно, также и в работе системы репарации неспаренных оснований (исправляет ошибки репликации ДНК и неправильную репарацию двунитевых разрывов ДНК) - см. раздел 3.11.3.
Помимо контроля повреждений ДНК и поддержания целостности генома BRCA1 выполняет и ряд других функций. Так, он связывает рецептор эстрогенов и репрессирует его транскрипционную функцию, сдерживая, таким образом, избыточную пролиферацию клеток молочной железы и других эстроген-зависимых органов, в частности при половом созревании и беременности. Кроме того, BRCA1, взаимодействуя с компонентами центросом (Nm23 и др.), принимает участие в обеспечении правильной сегрегации хромосом во время митоза.
Исходя из столь многочисленных функций BRCA1, становятся понятными последствия его инактивации. В клетках с дефектным BRCA1 наблюдается сильная генетическая нестабильность, т.е. повышение частоты возникновения спонтанных или индуцированных мутагенами генетических изменений - генных мутаций, хромосомных транслокаций, анеуплоидии и т.д. Кроме того, отменяется сдерживание пролиферации эстроген-зависимых клеток, что и объясняет, очевидно, возникновение опухолей именно молочной железы и яичника.
Функции белка BRCA2 изучены хуже. Как и BRCA1 он обладает репарационными и транскрипционными активностями. Связывая Rad51 (гомолог бактериального белка RecA), BRCA2 увеличивает его способность катализировать рекомбинации ДНК, обеспечивающие репарацию двунитевых разрывов ДНК. Транскрипционная функция BRCA2 связана, очевидно, со способностью рекрутировать P/CAF (p300/CBP Associated Factors), ацетилирующие гистоны и ремоделирующие хроматин. Однако физиологические гены-мишени BRCA2 пока не идентифицированы. Тем не менее о важности транскрипционной активности BRCA2 для его супрессорной функции может свидетельствовать тот факт, что обнаруживаемые в опухолях молочной железы мутации поражают именно транскрипционный домен. У мышей гомозиготный нокаут резко уменьшает жизнеспособность эмбрионов, а у выживших животных развиваются злокачественные тимомы. На клеточном уровне инактивация BRCA2 приводит к гиперчувствительности к различным генотоксическим агентам (УФ- и g-облучению, химическию мутагенам), повышению частоты встречаемости незарепарированных двунитевых разрывов ДНК и различных перестроек хромосом. Механизмы специфического возникновения у пациентов с герминальными мутациями BRCA2 опухолей молочной железы, яичника и простаты пока не установлены.
3.11.3. MSH2, MSH6, MLH1 и PMS2 - компоненты систем репарации неспаренных оснований ДНК
Риск развития новообразований значительно повышается и при врожденных дефектах системы репарации неспаренных оснований (mismatch repair), исправляющей главным образом ошибки репликации ДНК и неточности репарации двунитевых разрывов. В результате таких ошибок и потери комплементарности нитей ДНК возникают петли, которые распознаются комплексами белков MSH2/MSH6 или MSH2/MSH3 (они отличаются по способности узнавать разные типы петель, образующиеся при замене оснований, инсерциях и делециях). Эти комплексы рекрутируют к местам с нарушенной структурой ДНК комплексы белков MLH1/PMS2 или MLH1/MLH3, которые, в свою очередь, привлекают экзо- и эндонуклеазы, осуществляющие эксцизию аномального фрагмента ДНК, а также факторы репликации (PCNA, ДНК-полимеразы), обеспечивающие застройку бреши и восстановление нормальной структуры ДНК.
Врожденные гетерозиготные мутации по меньшей мере четырех из компонентов этой системы - MSH2, MLH1, MSH6 и PMS2 - вызывают синдром Линча. Главной чертой этого синдрома является развитие в молодом возрасте опухолей толстой кишки (так называемый наследственный неполипозный колоректальный рак) и/или опухолей яичника. Преимущественное возникновение опухолей кишечника, вероятно, связано с высочайшим пролиферативным потенциалом клеток на дне кишечных крипт, что естественно ведет и к более частому появлению ошибок репликации, которые должны исправляться именно системами репарации неспаренных оснований. Естественно, что бурно размножающиеся полустволовые (амплифицирующие) клетки кишечного эпителия накапливают необходимый для развития опухолей набор мутаций быстрее, чем медленно размножающиеся клетки.
Возникновение опухолей при дисфункции MSH2, MLH1, MSH3 или PMS2 связано, очевидно, с повышенной вероятностью мутаций в протоонкогенах и опухолевых супрессорах. Действительно, при мутациях гена MSH2 или MLH1 частота точечных мутаций во всех локусах увеличивается на 1-2 порядка, а в наследственных колоректальных раках, как правило, обнаруживаются точечные мутации в генах b-катенина, АРС, TbR-II, Smad2, Smad4 и т.д., которые, по-видимому, и являются причиной развития новообразований. Маркером инактивации любого из генов репарации неспаренных оснований является легко выявляемая нестабильность микросателлитных последовательностей ДНК. Нарушения функции генов MSH2, MLH1, MSH3, MSH6 и PMS2, приводящие к нестабильности микросателлитов, характерны и для некоторых форм спорадических (ненаследственных) опухолей: они обнаруживаются в 13-15% опухолей толстой кишки, рака желудка и эндометрия, но значительно реже (<2%) в других новообразованиях.
Описаны единичные случаи герминальных мутаций обоих аллелей гена MLH1, которые приводили к развитию еще во внутриутробном возрасте лимфосарком, лейкозов и нейрофиброматоза. Это объясняется видимо тем, что при полной инактивации системы репарации ошибок репликации ДНК и бурном размножении в эмбриогенезе клеток всех тканей, необходимое для образования опухоли количество мутаций успевает накопиться в каких-то клетках задолго до рождения, тогда как при гетерозиготных мутациях темп мутирования ниже и накопление мутаций до критического уровня продолжается в интенсивно размножающихся клетках взрослого организма. С этой точки зрения пока непонятно, почему у мышей как с гетерозиготным, так и с гомозиготным нокаутом гена MSH2 или гена MLH1, также развиваются лимфомы и саркомы, а не опухоли кишечника. (Впрочем, следует заметить, что мыши сильно отличаются от человека и по типу спонтанно развивающихся опухолей: у человека большую часть новообразований представляют различные формы рака, возникающие из эпителиоцитов, тогда как у мышей такие опухоли достаточно редки, а возникают, как правило, лимфомы и саркомы). Природу таких различий еще предстоит выяснить.
3.11.4.Компоненты системы эксцизионной репарации ДНК и пигментная ксеродерма
Система эксцизионной репарации узнает и исправляет сшивки оснований (тиминовые димеры и др.), образующиеся, например, после УФ-облучения или оксидативного стресса. Она включает множество компонентов. Распознавание тиминовых димеров осуществляется белковым комплексом XPC-hHR23, который вызывает рекрутирование к месту повреждения фактора TFIIH - сложного белкового комплекса, состоящего из 9 субъединиц и обладающего разными активностями, в том числе хеликазной и транскрипционной. Привлеченный фактор TFIIH катализирует раскрытие поврежденного участка ДНК и способствует сборке репарационного комплекса. Затем к дефектному участку последовательно рекрутируются белки XPG, XPA, комплекс RPA и, наконец, белки XPF-ERCC1, являющиеся эндонуклеазами. Именно они и осуществляют эксцизию поврежденного участка ДНК (обычно вырезается 24-32 нуклеотида) и инициируют застройку бреши по неповрежденной матрице и восстановление нормальной структуры ДНК.
Герминальные гетерозиготные мутации компонентов системы эксцизионной репарации, в частности генов XPA, XPB, XPC, XPD, XPF, XPG, ведут к возникновению пигментной ксеродермы - наследственного заболевания, характеризующегося повышенной чувствительностью к ультрафиолетовому облучению и развитием множественных опухолей кожи на местах, подвергающихся солнечному облучению. Интересно, что, несмотря на участие эксцизионной репарации в исправлении дефектов, вызванных не только УФ-облучением, но и мутагенами/канцерогенами, частота возникновения других форм опухолей при пигментной ксеродерме почти не увеличивается. При этом у трансгенных мышей с аналогичными дефектами системы эксцизионной репарации отмечается повышение частоты индукции новообразований химическими канцерогенами. Преимущественное возникновение у пациентов с пигментной ксеродермой исключительно опухолей кожи может указывать на незначительную роль химических факторов, загрязняющих окружающую среду, в развитии опухолей внутренних органов у человека.
ЗаключениеК настоящему времени идентифицировано несколько десятков генов,
инактивация которых приводит к развитию новообразований. Большинство
из них, регулируя клеточный цикл, апоптоз или репарацию ДНК, предотвращают
накопление в организме клеток с генетическими и некоторыми другими
аномалиями. Выявлены опухолевые супрессоры и с другими функциями,
в частности, контролирующие морфогенетические реакции клетки и
ангиогенез. Обнаруженные гены далеко не исчерпывают список существующих
опухолевых супрессоров. Уже сейчас в хромосомах человека картировано
более сотни участков, закономерно делетирующихся при различных
новообразованиях и, следовательно, содержащих потенциальные опухолевые
супрессоры. Их идентификация, вероятно, приведет к обнаружению
и других путей подавления опухолевого роста. В ближайшем будущем
можно ожидать также успехов в прикладном использовании знаний
об опухолевых супрессорах. Здесь следует надеяться, во-первых,
на разработку высокоточных методов диагностики наследственных
синдромов, предрасполагающих к развитию новообразований, а во-вторых,
на создание принципиально новых методов терапии злокачественных
новообразований, основанных на модификации сигнальных путей, контролируемых
опухолевыми супрессорами.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
1. Копнин Б.П. Мишени действия онкогенов и опухолевых супрессоров: ключ к пониманию базовых механизмов канцерогенеза. Биохимия, 2000, 65, 5-33.
2. Чумаков П.М. Функция гена р53: выбор между жизнью и смертью. Биохимия, 2000, 65, 34-47.
3. The Genetic Basis of Human Cancer. Eds Vogelstein B., Kinzler, K.W. McGraw Hill, New York, 1998.
4. Gray, J. W. & Collins, C. Genome changes and gene expression in human solid tumors. Carcinogenesis, 2000, 21, 443-452.
5. Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell, 2000, 100, 57-70.
6. Levine A. J. The tumour suppressor genes. Annu. Rev. Biochem., 1993, 62, 623-651.
7. Weinberg R.A. The molecular basis of oncogenes and tumor suppressor genes. Ann. N.Y. Acad. Sci., 1995, 758, 331-338.
8. Hooper M.L. Tumour suppressor gene mutations in humans and mice: parallels and contrasts. EMBO J., 1998, 17, 6783-6789.
9. Ghebranious N., Donehower L.A. Mouse models in tumor suppression. Oncogene, 1998, 17, 3385-3400.
10. Knudson, A. G. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA, 1971, 68, 820-823.
11. Grana X., Garriga J., Mayol X. Role of the retinoblastoma protein family, pRb, p107 and p130 in the negative control of cell growth. Oncogene, 1998, 17, 3365-3383.
12. Lipinski M.M., Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene, 1999, 18, 7873-7882.
13. Harbour J.W., Dean D.C. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev., 2000, 14, 2393-2409.
14. Paggi M.G., Giordano A. Who is the boss in the retinoblastoma family? The point of view of Rb2/p130, the little brother. Cancer Res., 2001, 61, 4651-4654.
15. Vogelstein B., Lane D., Levine A. Surfing the p53 network. Nature, 2000, 408, 307-310.
16. Levine A.J. p53, the celular gatekeeper for growth and division. Cell, 1997, 88, p.323 - 331.
17. Woods, D. B. & Vousden, K. H. Regulation of p53 function. Exp. Cell Res., 2001, 264, 56-66.
18. Prives, C. and Hall, P.A. The p53 pathway. J. Path., 1999, 187, 112-126.
19. Sionov R.V., Haupt, Y. The cellular response to p53: the decision between life and death. Oncogene, 1999, 18, 6145 - 6157.
20. Yang A, McKeon F. p63 and p73: p53 mimics, menaces and more. Nat. Rev. Mol. Cell. Biol., 2000, 1, 199-207.
21. Sherr C. J. & Weber, J. D. The ARF/p53 pathway. Curr. Opin. Genet. Dev., 2000, 10, 94-99.
22. Ruas M., Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochem. Biophys. Acta, 1998, 1378, F115-F177.
23. Sherr C. J. Parsing Ink4a/Arf: "pure" p16-null mice. Cell, 2001, 106, 531-534.
24. Maehama, T. & Dixon, J. E. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol., 1999, 9, 125-128.
25. Bonneau, D. & Longy, M. Mutations of the human PTEN gene. Hum. Mutat., 2000, 16, 109-122.
26. Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim.Biophys. Acta, 1997, 1332, F127-F147.
27. Polakis P. The oncogenic activation of b-catenin. Curr. Opin. Genet. Dev., 1999, 9, 15-21.
28. Polakis P. Wnt Signaling and cancer. Genes Dev., 2000, 14, 1837-1851.
29. Polakis P. More than one way to skin a catenin. Cell, 2001, 105, 563-566.
30. Taipale J., Beachy P.A. The Hedgehog and Wnt signaling pathways in cancer. Nature, 2001, 411, 3503-354.
31. Bienz M., Clevers H. Linking colorectal cancer to Wnt signaling. Cell, 2000, 103, 311-320.
32. Massague J., Blain S.W., Lo R.S. TGFb signaling in growth control, cancer, and heritable disorders. Cell, 2000, 103, 295-309.
33. Massague J. How cells read TGF-? signals. Nature Rev. Mol. Cell Biol., 2000, 1, 169-178.
34. Blagosklonny M.V. Do VHL and HIF-1 mirror p53 and Mdm-2? Degradation-transactivation loops of oncoproteins and tumor suppressors. Oncogene, 2001, 20, 395-398.
35. Gutmann D.H., Hirbe A.C., Haipek C.A. Functiomal analysis of neurofibromatosis 2 (NF2) missense mutations. Hum. Mol.Genet., 2001, 10, 1519-1529.
36. Kinzler, K. W. & Vogelstein, B. Gatekeepers and caretakers. Nature 386, 1997, 761-763.
37. Zhou B.-B.S., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature, 2000, 408, 433-439.
38. Lengauer C., Kinzler K.W., Vogelstein B. Genetic instabilities in human cancers. Nature, 1998, 396, 643-649.
39. Ponder B.A.J. Cancer genetics. Nature, 2001, 411, 336-341.
40. Shiloh, Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu. Rev. Genet., 1997, 31, 635-662.
41. Kastan M.B., Lim D.S. The many substrates and functions of ATM. Nature Rev. Mol. Cell. Biol., 2000, 1, 179-186.
42. Walworth N.C. Cell-cycle checkpoint kinases: checking in on the cell cycle. Curr. Opin. Cell Biol., 2000, 12, 697-704.
43. Welsh P.A., King M.-C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol.Genet., 2001, 10, 705-713.
44. Zheng L., Li S., Boyer T.G., Lee W.-H. Lessons learned from BRCA1 and BRCA2. Oncogene, 2000, 19, 6159-6175.
45. Wang Q., Zhang H., Fishel R., Greene M.I. BRCA1 and cell signaling. Oncogene, 2000, 19, 6152-6158.
46. Kolodner R.D., Marsischky G.T. Eukaryotic DNA mismatch repair. Curr. Opin.Genet. Dev., 1999, 9:89-96.
47. de Laat W.L., Jaspers N.G.J., Hoeijmakers J.H.J. Molecular mechanism of nucleotide excision repair. Gen. Dev., 1999, 13, 768-785.
48. Lehman A.R. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Gen. Dev., 2001, 15, 15-23.