Онкология-
Опухолевые супрессоры и мутаторные гены
Б.П.Копнин
Российский онкологический научный центр им. Н.Н.Блохина РАМН, Москва
источник RosOncoWeb.Ru
01 02 03 04 05 06 073.6. Е-кадгерин, АРС и аксин - супрессоры, контролирующие сигнальный путь ?-катенин/Cdk/pRb
Наследственные и спорадические опухоли желудочно-кишечного тракта довольно часто ассоциированы с мутациями в генах Е-кадгерина, ?-катенина, АРС и аксина (Табл. 1). Так, гемизиготные герминальные мутации Е-кадгерина обусловливают развитие наследственных форм рака желудка. Врожденные мутации одного из аллелей гена ?-катенина или гена APC ведут к развитию семейного аденоматозного полипоза кишечника (у большинства пациентов наблюдаются мутации APC, реже встречаются мутации ?-катенина). В основе онкогенного эффекта всех этих мутаций лежит повышение транскрипционной функции протоонкобелка ?-катенина, вызывающего активацию циклинзависимых киназ и инактивирующего тем самым супрессорную функцию pRb (см. раздел 3.2.2). Такая функция ?-катенина обусловлена его способностью транслоцироваться в ядро, связываться с транскрипционными факторами семейства TCF/LEF и активировать гены, имеющие в своем составе TCF/LEF-респонсивные элементы, в частности ген циклина D1 и онкоген MYC.
Е-кадгерин - представитель семейства трансмембранных гликопротеинов, осуществляющий адгезионные межклеточные контакты типа "зона слипания" (zona adhaerence). Его внутриклеточный домен связывается с рядом белков, в первую очередь с b-катенином. В этом случае, происходит переключение функции ?-катенина: он перестает функционировать в качестве транскрипционного фактора, а взаимодействует с актиновыми микрофиламентами и участвует в регуляции реорганизации цитоскелета. Характерные для опухолей желудочно-кишечного тракта, рака молочной железы, яичника и ряда других новообразований изменения гена Е-кадгерина (мутации, делеции, метилирование) вызывают потерю экспрессии белка или изменение его локализации в клетке, что имеет два важных для онкогенеза последствия. Во-первых, нарушаются межклеточные контакты и морфогенетические реакции клетки, а, во-вторых, происходит накопление ?-катенина в ядре и повышение его транскрипционной активности (Рис. 6). В результате стимулируется размножение клеток и увеличивается их способность к инвазии. Восстановление экспрессии Е-кадгерина в раковых клетках с помощью введения генно-инженерных конструкций вызывает резкое замедление пролиферации и переход от инвазивного к неинвазивному фенотипу. Причем рост-супрессирующий эффект Е-кадгерина обусловливается его способностью связывать и секвестрировать b-катенин, и не зависит от того, происходит ли при этом восстановление межклеточных контактов.
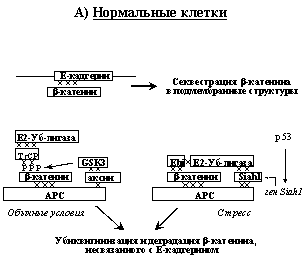
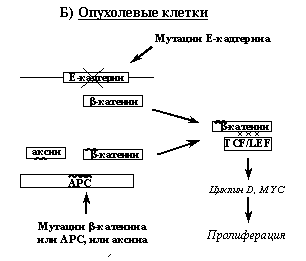
Рис. 6. Регуляция транскрипционной активности ?-катенина и ее нарушения в опухолевых клетках (объяснения в тексте).
В несвязанном с Е-кадгерином состоянии ?-катенин очень нестабилен. Ключевую роль в регуляции его времени жизни и транскрипционной активности играют опухолевые супрессоры APC и аксин (Рис. 6А). АРС связывает одновременно и b-катенин и аксин, в результате чего образуется сложный комплекс, к которому рекрутируется GSK-3b (киназа-3b гликогенсинтетазы). Фосфорилируя определенные сериновые остатки ?-катенина, GSK-3b индуцирует связывание его с Е2-убиквитин-лигазой. Убиквитинированный b-катенин направляется в протеосомы, где происходит его деградация. При стрессах, вызывающих активацию р53, включается дополнительный механизм деградации ?-катенина (Рис. 6А). В этом случае происходит повышение экспрессии белка Siah1, который рекрутирует другой тип комплексов Е2-убиквитин-лигазы также и к нефосфорилированному по N-концу b-катенину. Таким образом достигается, по-видимому, более эффективная деградация несвязанного с кадгеринами ?-катенина.
Вероятно, при онкогенных мутациях Е-кадгерина свободного ?-катенина становится так много, что системы его деградации не справляются и часть ?-катенина оказывается в ядре, где он проявляет свою транскрипционную активность (Рис. 6Б). В клетках новообразований часто реализуется и другой путь повышения транскрипционных активностей ?-катенина, связанный с нарушениями работы систем его деградации. Так, в клетках опухолей толстой кишки, печени, простаты часто обнаруживаются либо мутации ?-катенина в участках, которые фосфорилируются GSK-3b и взаимодействуют с убиквитин-лигазой, либо мутации опухолевого супрессора APC, нарушающие его взаимодействие с b-катенином. либо (значительно реже) мутации аксина, отменяющие его связывание с АPC или GSK-3b. (При этом мутации APC блокируют работу обоих систем деградации ?-катенина, тогда как указанные мутации самого ?-катенина или аксина нарушает только одну из этих систем). В результате всех этих событий b-катенин активирует транскрипцию гена циклина D и протоонкогена MYC, что вызывает стимуляцию клеточной пролиферации.
Е-кадгерин и АРС являются классическими опухолевыми супрессорами - у пациентов с семейными формами соответственно рака желудка и аденоматозного полипоза кишечника в опухолевых клетках наблюдается инактивация обоих аллелей данных генов. Произошедшие еще в половой клетке наследуемые изменения одного из аллелей представляют собой точечные инактивирующие мутации или микроделеции. Второй аллель инактивируется уже в соматической клетке. Подавление активности второго аллеля гена Е-кадхерина нередко связано с его метилированием, а второй аллель гена АРС чаще всего инактивируется в результате делеции участка длинного плеча хромосомы 5, содержащего данный ген, или утраты всей хромосомы 5. Ключевая роль герминальных мутаций APC в генезе аденоматозного полипоза толстой кишки подтверждается тем, что у трансгенных мышей с гомозиготным нокаутом этого гена также развиваются множественные полипы, причем не только в толстом, но и в тонком кишечнике.
Недавно выявлена еще одна важная биологическая функция опухолевого супрессора АРС. Во время митоза он связывается с белками кинетохора и участвует в организации веретена деления. При инактивации APC нарушается взаимодействие микротрубочек с кинетохором, результатом чего является частые ошибки сегрегации хромосом, т.е. нестабильность генома. Это, вероятно, может служить объяснением более частой встречаемости мутаций APC по сравнению с мутациями ?-катенина: помимо более эффективного блокирования работы систем деградации ?-катенина они ведут также и к генетической нестабильности, что придает клеткам дополнительные селективные преимущества.
3.7. Компоненты сигнальных путей TGFb-SmadКак опухолевые супрессоры классифицированы и некоторые компоненты сигнальных путей, регулируемых TGF-?. Этот цитокин, в зависимости от типа клеток-мишеней и их микроокружения, может вызывать остановку размножения, стимуляцию дифференцировки, а в некоторых случаях и апоптоз. Свои антипролиферативные и дифференцировочные эффекты (они наблюдаются в нормальных эпителиальных, эндотелиальных и гемопоэтических клетках) TGF-? реализует по следующему механизму. Его связывание с RII-субъединицей рецептора, обладающей серин-треонин киназной активностью, вызывает рекрутирование и фосфорилирование второй субъединицы рецептора - RI, также являющейся серин-треониновой киназой. Основной мишенью киназы Tb-RI являются, в зависимости от типа клеток, белки Smad2 или Smad3 - так называемые рецепторные Smad. Их фосфорилированные формы образуют комплекс с белком Smad4, который транспортируется в ядро и, образуя еще более сложные комплексы с другими транскрипционными кофакторами, функционирует в качестве активаторов транскрипции одних генов и репрессоров других генов. В частности, он репрессирует ген MYC и транс-активирует гены ингибиторов циклинзависимых киназ (CKIs) p15Ink4b, p27Kip1 и p21Waf1/Cip1, что ведет к ингибированию функции Cdk4, Cdk2 и остановке клеточного цикла в G1. При этом, репрессия MYC играет ключевую роль в проведении антипролиферативного сигнала TGF-?, так как именно она "разрешает" стимуляцию транскрипции генов CKI комплексом Smad2(3)/Smad4/Sp1 (предполагается, что Myc закрывает места посадки этого комплекса в генах p15Ink4b, p27Kip1 и p21Waf1/Cip1). При гиперэкспрессии онкогена МYC, которая характерна для многих новообразований человека, TGF-? не способен вызвать понижение его экспрессии, достаточное для "разрешения" стимуляции транскрипции генов CKI, и в результате TGF-? не оказывает антипролиферативного действия.
Инактивирующие мутации компонентов этого сигнального пути, а
именно рецептора TGF-? (TbR-II), Smad2 и Smad4 характерны для
опухолей толстого кишечника, рака поджелудочной железы, желчного
пузыря, легкого, а также некоторых других новообразований. Герминальные
мутации в одном из аллелей генов TbR-II или Smad4 ассоциированы
с развитием семейных форм рака толстого кишечника и желудка. Интересно,
что у трансгенных мышей с повреждением одного из аллелей гена
TbR-II, Smad2 или Smad4 частота развития опухолей не повышается,
однако гетерозиготная инактивация одновременно и гена Smad4 и
гена APC резко увеличивает вероятность развития инвазирующих опухолей
кишечника. Сходная картина наблюдается у мышей с нокаутом одного
из аллелей гена Smad3. В этом случае наблюдается развитие в молодом
возрасте множественных метастазирующих колоректальных карцином.